
Abstract
It remains a challenge to identify the active sites of bismuth catalysts in the electrochemical CO2 reduction reaction. Here we show through in situ characterization that the activation of bismuth oxyhalide electrocatalysts to metallic bismuth is guided by the halides. In situ X-ray diffraction results show that bromide promotes the selective exposure of planar bismuth surfaces, whereas chloride and iodide result in more disordered active sites. Furthermore, we find that bromide-activated bismuth catalysts outperform the chloride and iodide counterparts, achieving high current density (>100 mA cm–2) and formic acid selectivity (>90%), suggesting that planar bismuth surfaces are more active for the electrochemical CO2 reduction reaction. In addition, in situ X-ray absorption spectroscopy measurements reveal that the reconstruction proceeds rapidly in chloride-activated bismuth and gradually when bromide is present, facilitating the formation of ordered planar surfaces. These findings show the pivotal role of halogens on selective facet exposure in activated bismuth-based electrocatalysts during the electrochemical CO2 reduction reaction.

Similar content being viewed by others


Main
The electrocatalytic conversion of CO2 into chemicals and fuels is considered as a powerful tool to store intermittent renewable energy in chemical bonds1,2,3,4. However, there remain several challenges that need to be faced before the full potential of electrocatalytic CO2 conversion can be used. Among these challenges, a lot of research has been devoted to the optimization of three key descriptors of catalysis, that is, selectivity, activity and stability5,6,7,8,9. Despite the fact that C2+ products (for example ethylene and ethanol) have a greater industrial value than C1 products (for example carbon monoxide (CO) and formic acid (HCOOH)), the complicated reaction mechanism for C2+ products hampers the development of selectivity and activity10. Furthermore, recent techno-economic analysis suggests that the synthesis of CO or formic acid as a base chemical is economically and practically more feasible due to the high production rate and superior adaptability to future industrial implementation compared to C2+ products11,12,13. In numerous chemical processes, including electrowinning, leather tanning and aviation de-icing, formic acid has been extensively investigated as chemical feedstock14,15,16 with a sizeable economic market17,18.
Over the past decades, efforts have been aimed at the optimization of electrocatalysts for HCOOH production19. Post-transition-metal-based electrocatalysts, like tin (Sn), indium (In), bismuth (Bi) and lead (Pb), have been extensively investigated as the typical catalysts for selective CO2 to HCOOH electrochemical conversion due to their strong adsorption of the key intermediates towards formic acid production and suppression of the competing hydrogen evolution reaction15,20,21,22. Among the post-transition metals, Sn and In have been studied extensively due to their low costs, relative abundance and easy processability. Due to toxicity reasons, Pb is not vastly studied for electrochemical CO2 conversion, but has shown promise in combination with Cu for selective CO production23. Bi-based electrocatalysts for CO2 conversion were first reported in 1995, but since then research into Bi-based electrocatalysts has stagnated24,25. Recently, Bi-based electrocatalysts have received increasing attention because of the low toxicity and high abundance of Bi (refs. 26,27). However, there is still a lot of room for improvement in terms of activity, selectivity and stability of Bi-based electrocatalysts28.
Various strategies, such as the use of nanostructures, defects and selective facet exposure, have been investigated to boost the selectivity and activity of electrocatalysts. However, the actual active site, stability and reaction mechanism under operating conditions are still up for debate29. By employing in situ attenuated total reflectance infrared spectroscopy to observe the unaltered oxide layer on the Bi surface, Pander et al. concluded that Bi is an oxide-independent electrocatalyst, although other researchers found the Bi–O interaction to be crucial for the effectiveness of Bi-based electrocatalysts in the electrochemical CO2 reduction reaction (eCO2RR)30,31. Along with the debate regarding the participation of oxide species, density functional theory findings have suggested that the basal Bi(003) plane is less active than the edge plane Bi(012) in HCOOH formation31, while some other work stated that basal sites are more active15,32,33,34. Furthermore, it has been reported that the use of layered metallic Bi electrocatalysts is beneficial in the eCO2RR since the layered structure often results in improved performance35,36,37,38,39. Identification of the active sites and disclosure of the restructuring during operation are crucial in the study of the Bi-catalysed eCO2RR in order to logically design a catalyst with improved activity, selectivity and stability to satisfy the requirement of practical applications. X-ray radiation is ideally suited for this purpose, because it can probe structural features over multiple length scales, depending on the X-ray technique of choice. However, online monitoring of structural changes and distinguishing between the sites responsible for the CO2 conversion reaction require dedicated in situ X-ray characterization cells due to the attenuation of X-rays by the aqueous electrolyte40,41.
In this Article, layered {001}-oriented Bi oxyhalide (further denoted as BiOX, with X = Cl, Br or I) nanoplatelets were synthesized and used as a platform to in situ monitor the structure and active sites of Bi-based electrocatalysts in the eCO2RR. Through in situ X-ray diffraction (XRD) and in situ Raman spectroscopy measurements, we followed the dynamics of the in situ transformation of the prepared BiOX catalyst materials into activated metallic Bi electrocatalysts. It was found that the in situ-activated Bi electrocatalysts selectively exposed specific facets during catalysis, guided by the choice of halide: Br– promoted Bi(003) exposure and Cl– resulted in dominant Bi(012) facets, whereas I– created a mixture. Furthermore, we linked the facet exposure to catalytic performance. BiOBr (BOB) exhibited a maximum HCOOH selectivity of 91% with a current density of 148 mA cm–2 at –1.05 V versus reversible hydrogen electrode (RHE). At a similar potential of –1.09 V versus RHE, BiOCl (BOC) displayed 69% HCOOH Faradaic efficiency (FE) at 88 mA cm–2 current density. BiOI (BOI) demonstrated a HCOOH selectivity of 76% at –1.08 V versus RHE with a current density of 95 mA cm–2, between BOB and BOC. This suggests that the in situ-formed basal Bi(003) facet is more catalytically active than the stepped Bi(012) site in the eCO2RR. In addition, in situ X-ray absorption spectroscopy (XAS) measurements revealed that the reconstruction of BOC proceeded rapidly after reduction onset, as indicated by the short-lived transition state, while the transition state in BOB and BOI persisted longer. This implies that halogens in BiOX affect the selective facet exposure of the formed Bi during catalysis by tuning the reconstruction rate. This work provides further insight into the active sites and structural evolution of Bi-based electrocatalysts in electrocatalytic CO2 conversion and paves the way for the rational design of electrocatalysts for liquid fuel production through in situ characterization studies.
Results
Structural characterization of bismuth oxyhalide nanoplatelets
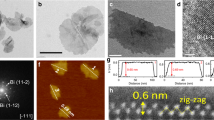
Figure 1a depicts a typical BiOX layered structure. Through weak van der Waals interactions, the neighbouring halogen layers in the BiOX stack and the [Bi2O2]2+ slab are sandwiched between two layers of halogen atoms42. The studied BiOBr (BOB), BiOCl (BOC) and BiOI (BOI) catalysts were prepared through hydrothermal synthesis (see Methods). To determine the crystal phase and electronic structure of the obtained materials, XRD and X-ray absorption near edge spectroscopy (XANES) were employed. As shown in Fig. 1b, a typical layered BiOCl structure is observed with (001), (002) and (003) planes dominating the XRD pattern of BOC. The XRD patterns for BOB and BOI show similar layered structures, with the reflections slightly shifted to smaller diffraction angles due to the size of the halogen. Similar to BOC, a series of {001} facets dominates the XRD patterns, with negligible reflections of other facets. In Fig. 1c, the XANES spectra of the studied BOB, BOI and BOC are compared with those of reference metallic Bi powder and Bi2O3. BOB, BOI and BOC show a similar shape of XANES spectra, suggesting a shared electronic structure for Bi. It is clear that the Bi L3-edge position of the studied materials differs from that of metallic Bi, but closely resembles that of Bi2O3. This is further confirmed by the first derivative of the XANES spectra (Supplementary Fig. 1), showing that the studied BOB, BOI and BOC have a similar oxidation state to Bi2O3, suggesting the presence of Bi3+.

a, Representative schematic crystal structure of BiOX (exemplified by BiOI). b, XRD patterns of the studied BOC, BOB and BOI electrocatalysts. c, XANES spectra of BOB, BOC and BOI compared with those of the reference compounds Bi powder and Bi2O3, plotted as normalized absorbance (in arbitrary units, a.u.) versus energy. d,e, Magnified HAADF-STEM image of BOI and the corresponding STEM-EDS elemental map, revealing the distribution of Bi, O and I. f, AFM image (corresponding thickness profile in Supplementary Fig. 10), in which the colour scale denotes sample height (in nm).
To gain more insights into the structure of the prepared Bi-based catalysts, morphological structure and elemental distribution were investigated using scanning electron microscopy (SEM), high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) and coupled STEM energy dispersive X-ray spectroscopy (STEM-EDS) measurements. The representative SEM images in Supplementary Fig. 2 show that all BiOX materials have a layered morphology, which is consistent with the XRD results. BOB has an irregular film structure that ranges in lateral size from 200 nm to 3 µm. This is similar to the morphology of BOI, which also exhibits an irregular film structure with smaller lateral dimensions, ranging from 100 nm to 300 nm. In BOC, uniform squares are observed with edge length around 1 µm. We hypothesize that BOC grows more isotropically due to the stronger Bi–Cl bonds43, compared to the Bi–Br and Bi–I bonds, similar to the growth mechanism for other two-dimensional compositions44. The lower bond energy makes it energetically favourable for BOB and BOI to grow anisotropically based on the layered crystal structure, whereas in BOC additional Bi–Cl bonds are created by growing thicker two-dimensional structures. The high-resolution HAADF-STEM image of BOI is shown in Supplementary Fig. 3, confirming its crystalline structure, whereas the HAADF-STEM image (Fig. 1d), along with the corresponding combined elemental maps (Fig. 1e) and EDS spectrum (Supplementary Fig. 4), confirms the homogeneous distribution of Bi, O and I in the particles. From the high-resolution HAADF-STEM images and the corresponding Fourier transform (FT) patterns of BOB particles (Supplementary Fig. 5), two different lattices could be observed in the bulk and edge area of the BOB particles. These lattices were identified as typical BiOBr and Bi2O2CO3, respectively. The presence of Bi2O2CO3 at the surface is attributed to inevitable air exposure during sample transport. Similar to BOI, uniform elemental distribution of Bi, O and Br/Cl is observed in BOB/BOC, respectively (Supplementary Figs. 6–9). The morphology and thickness of the studied materials were further investigated using atomic force microscopy (AFM). In BOB (Supplementary Fig. 10), overlapping irregular layers are observed with thicknesses ranging from 70 to 90 nm, confirming the layered structure. BOI has a similar irregular layered structure, but its thickness is estimated to be around 40–50 nm (Fig. 1f and Supplementary Fig. 10c). BOC, on the other hand, is much thicker than both BOB and BOI, with a regular square shape and an approximate thickness of 600 nm (Supplementary Fig. 11).
Halide influence on eCO2RR performance
The electrocatalytic performance of the BiOX electrocatalysts in the eCO2RR was first explored using an H-type cell with a typical three-electrode configuration in 0.1 M KHCO3 aqueous electrolyte (see Methods). This concentration for the electrolyte was chosen in this work because it allowed for direct comparison between different studies into the structure–performance relationships for electrocatalysts45 and the potentials reported were automatically corrected to account for the uncompensated solution resistance (iR)46. Linear sweep voltammetry measurements were employed to estimate the activity of the studied catalyst materials. In Supplementary Fig. 12, the current densities for these three catalysts in the CO2-saturated electrolyte are plotted and it is observed that all current densities are larger than the corresponding current density in the N2 atmosphere, indicating their intrinsic activity for the eCO2RR. Besides, BOB has a larger current density difference in CO2 and N2 than BOI and BOC, while the difference between BOI and BOC is minor, indicating that the activity of BOB in the eCO2RR is better than that of BOI and BOC. To analyse the selectivity of different eCO2RR products under various potentials, we have applied stepped-potential electrolysis across a potential range from –0.85 to –1.25 V versus RHE and detected the gaseous products with online gas chromatography and liquid products with offline NMR after around 40 min of eCO2RR. We can see, from Supplementary Fig. 13, that the FE of BOB for HCOOH is higher than 80% over the entire potential region and it reached up to 96% from −0.95 to −1.15 V versus RHE in the H-type cell, with effective suppression of the hydrogen evolution reaction and CO production. BOI had a maximum HCOOH FE of 85% at −1.15 V versus RHE, slightly lower than BOB, while BOC displayed a maximum HCOOH FE of 81% at −1.05 V versus RHE. After prolonged operation (up to 8 h), the selectivity for all BiOX electrocatalysts investigated degraded to 60% (Supplementary Fig. 14) and the production of H2 increased over time.
To evaluate the electrocatalytic CO2 conversion performance of the as-prepared BiOX materials at current densities as close as possible to industrial relevance47, a gas diffusion electrode (GDE) flow cell was used with 1 M KHCO3 solution as the electrolyte. To account for the change in solution resistance, the potentials in the GDE cell were manually iR-corrected46. Within the GDE cell configuration (Supplementary Fig. 15), continuous gaseous CO2 feedstock could be supplied through the porous carbon support to the catalyst surface so that the gas solubility limitation in the aqueous electrolyte was circumvented. On the other side of the porous electrode, electrolyte flowed over the catalyst surface with the help of a peristaltic pump, hereby diluting the HCOOH accumulation at the catalyst surface and buffering the electrolyte pH change, in order to minimize the mass transfer limitations. The minimization of mass transfer limitations was confirmed by the fact that the current densities using the GDE flow cell were 60 times higher than using the H-type cell (Supplementary Figs. 12 and 16). Figure 2a,b indicates that not only does the selectivity for BOB reach a maximum around –1.05 V versus RHE, but also the production rate of BOB outperforms the other two compositions in the investigated potential range.

a,b, Comparison of FE and production rate of HCOOH in BOB, BOI and BOC. Error bars were obtained by measuring the liquid products in triplicate and the centre value for the error bars is the average of the three independent measurements. c, The corresponding partial current density in CO2-saturated 1 M KHCO3 electrolyte solution using a GDE flow cell. All potentials were iR-corrected (90%).
Current density is commonly used as the main indicator to evaluate catalyst activity. BOB has the highest total current density over the applied potential window of the studied catalysts, as shown in Supplementary Fig. 17. When solely HCOOH FE is considered, there is a large difference in partial current density between BOB, BOI and BOC (Fig. 2c). At an applied potential of −1.05 V versus RHE, a HCOOH partial current density of 148 mA cm–2 is achieved for BOB, whereas BOI and BOC only reach 95 and 88 mA cm–2 at −1.08 V versus RHE and −1.09 V versus RHE, respectively. Different morphological features are known to result in different catalytic behaviour and larger specific surface areas usually expose more active sites48. In order to accurately define the contribution of the nature and number of active sites during electrolysis, we evaluated the electrochemical surface area (ECSA) of the as-prepared and spent catalysts by measuring the double-layer capacitance (Cdl) in the non-Faradaic potential area in an H-type cell (Supplementary Figs. 18–22)49. Before catalysis, BOC had twice the double-layer capacitance of BOB and BOI, but after catalysis all investigated catalysts showed similar values. Considering that BOC resulted in lower selectivity and activity than BOB and BOI, we conclude that the larger ECSA did not contribute much to catalytic performance. Besides, the catalyst structures underwent reconstruction during catalysis, resulting in similar ECSA after catalysis. This is also supported by electrochemical impedance spectroscopy (Supplementary Fig. 23). It can be seen that BOB, BOC and BOI have a similar semicircle in the high frequency area at the open circuit potential (OCP), implying a similar charge transfer property. When a reaction potential was applied, they all formed a second semicircle, which is the signal of the generation of a second interface, suggesting the structure of the catalysts studied changed at such a potential. This can be further confirmed by their SEM images before and after catalysis (Supplementary Figs. 24–26).
Halide-dependent reduction through in situ Raman spectroscopy
Based on the observations above, we have used in situ techniques to monitor the structural evolution of the catalyst materials under reaction conditions and to elucidate the relationship between catalyst reconstruction and catalytic activity. In situ Raman spectroscopy measurements were performed at a rate of 10 s per spectrum. As shown in Supplementary Fig. 27a, BOB exhibits BiOBr characteristics at the start of the reaction (A1g Bi–Br band at 110 cm–1 and Eg Bi–Br band at 158 cm–1) and these characteristics all fade away after 80 s at a fixed applied potential of –1.15 V versus RHE (ref. 50). The peak at 384 cm–1 is attributed to the Eg and B1g bands produced by the motion of oxygen atoms51,52. The peak located at 478 cm–1 is related to the Bi–O vibration, which disappears at around 150 s (Supplementary Fig. 28) in BOB (ref. 53). Similarly, BiOI features (A1g Bi–I band at 107 cm–1 and Eg Bi–I band at 150 cm–1) are observed in BOI at the onset of catalysis, which disappear after 30 s of cathodic bias (Supplementary Figs. 27b and 29)54. The Bi–O band at 478 cm–1 in BOI also disappears at around 150 s. In BOC, BiOCl features (A1g Bi–Cl band at 145 cm–1 and Eg Bi–Cl band at 200 cm–1) fade away after 80 s of catalysis (Supplementary Fig. 27c), similar to BOB. We note that these vibrational features for the Bi–halide bonds confirm that Bi–Cl forms the strongest bond, supporting our hypothesis that the bond strength determines the growth dynamics of the BiOX structures. However, the Bi–O band persists in BOC even after 210 s (Supplementary Fig. 30), indicating that the reduction of BOC is hampered compared to those of BOB and BOI (ref. 55). This implies that reconstructions occur in the pristine Bi oxyhalides during catalysis and the conversions of BOB and BOI are easier than that of BOC under the same applied cathodic potential. The relatively poor reducibility of BOC can be attributed to the larger size and thickness than the other Bi oxyhalide nanoplatelets56. Due to detection limitations and a low signal-to-noise ratio, it is difficult to see other formed species, such as reaction intermediates, during catalysis using in situ Raman spectroscopy.
Halide-guided active site exposure through in situ XRD
Dynamic structural information was obtained by performing in situ XRD measurements to reveal the structure evolution during catalysis. The time-dependent in situ XRD pattern was collected every 3 min, except for the first two XRD patterns (that were collected each minute) in order to catch the small changes at the beginning of the reaction. In Fig. 3a and Supplementary Fig. 31a, it can be seen that BiOBr {001} facets dominate in BOB at the OCP, which quickly fade away within 7 min at an applied potential of −1.15 V versus RHE (intensity versus time trace in Fig. 3d), while reflections of metallic Bi (PDF # 00–004–1246) begin to appear after 1 min already with dominant arrangements of Bi(003) and Bi(006) at 22.4 and 45.8°, respectively. Another constant feature located at 44.3° is from the cell cover. Under the same conditions, distinct characteristic BiOI reflections can be seen in BOI (Fig. 3b and Supplementary Fig. 31b) at the start of the reaction, which also disappear in the first 4 min according to the intensity versus time trace (Fig. 3e). Simultaneously, Bi(003) and Bi(006) appear in the XRD patterns, with the same contributions from Bi(012), Bi(104) and Bi(110) facets located at 27.1, 37.9 and 39.6°, respectively. Furthermore, BOC exhibits BiOCl peaks at the start of the reaction, which gradually decline during catalysis (Fig. 3c and Supplementary Fig. 31c). Furthermore, no Bi(003) and Bi(006) reflections are observed for BOC after 40 min of cathodic bias, evidenced by the intensity versus time trace (Fig. 3f). These in situ time-dependent XRD measurements indicate that an applied potential of –1.15 V versus RHE is sufficient driving force to reduce BOB and BOI, whereas for BOC higher applied potentials are required.

a–c, Time-dependent in situ XRD patterns at a fixed potential of −1.15 V versus RHE of BOB (a), BOI (b) and BOC (c) in CO2-saturated 0.1 M KHCO3 solution, plotted in arbitrary units (a.u.) versus diffraction angle 2θ. d–f, Diffraction intensity of Bi(003) and Bi(012) facets as a function of reaction time of BOB (d), BOI (e) and BOC (f) in CO2-saturated 0.1 M KHCO3 solution. The purple and green shaded areas give the expected diffraction angles for the Bi(003) and Bi(006) reflections and Bi(012) reflections, respectively. The inserted structural models display the Bi(003) and Bi(012) facets from the top view.
Since the studied catalyst materials have shown potential-dependent catalytic performance (Fig. 2a), potential-dependent in situ XRD was also conducted to reveal the structure evolution as a function of applied potential, in which a diffraction pattern was collected every –0.20 V versus RHE. Figure 4a,d and Supplementary Fig. 32a illustrate the same trend as the result in time-dependent measurements, with BOB showing characteristic BiOBr reflections at the OCP and converting into metallic Bi at the cathodic potential onset of −0.15 V versus RHE. This is consistent with the cyclic voltammetry (CV) result (Supplementary Fig. 18), in which the two reduction peaks of Bi3+/Bi are located at potentials of −0.02 V versus RHE and −0.15 V versus RHE. The BiOBr reflections fully disappear at an applied cathodic bias that is a bit more negative than that given by the CV result (<–0.15 V versus RHE). A possible reason for this is that CV mainly measures the surface redox properties, whereas XRD detects structural changes of the bulk electrode. As the potential-dependent catalysis proceeds, Bi(003) and Bi(006) facets remain the main exposed facets for the BOB-derived electrocatalysts. Likewise, BOI also shows typical BiOI reflections at the OCP (Fig. 4b,e and Supplementary Fig. 32b), but with increasing negative potentials these peaks gradually decrease in intensity. Metallic Bi peaks already appear at a potential of −0.15 V versus RHE, which is also in agreement with the CV result for Bi3+/Bi reduction at this potential. Furthermore, this result indicates that the reducibility of BOI is higher compared to that of BOB, since the characteristic BOI reflections have faded out at a lower cathodic bias. A similar intensity of Bi(003), Bi(006) and Bi(012) facets is observed in BOI during catalysis, just as in the time-dependent in situ XRD measurements (Fig. 3b). This indicates that the activated Bi catalyst has the same conformation regardless of time- or potential-dependent electrocatalytic activation. Figure 4c,f and Supplementary Fig. 32c show the in situ XRD patterns of BOC at different potentials. BiOCl reflections are observed at the OCP and they gradually fade away when larger cathodic potentials are applied. The most striking difference between BOC and BOB/BOI is that the Bi(012) facet is the most pronounced in BOC during catalysis, along with the less pronounced Bi(104) and Bi(110) facets, while Bi(003) and Bi(006) are effectively suppressed. The reflections of BiOCl remain at the applied cathodic bias of −1.35 V versus RHE. This is consistent with the observation in the CV curve for BOC activation that the Bi3+/Bi reduction peak requires a more negative potential compared to BOB and BOI. As mentioned above, this could be due to the relatively large thickness of BOC, which normally requires more driving force to be fully reduced. The observed residue of BiOCl is attributed to the uneven catalyst layer on the glassy carbon wafer, as the drop-casting method was used to prepare the electrode. Results of the ex situ grazing incidence XRD (GIXRD, Supplementary Fig. 33) measurement, which was used to determine surface structure, confirm that all of the studied BiOX materials have been reduced to metallic Bi surfaces after catalysis. In addition, on the surface of the spent BOB, only Bi(003) can be observed, while on the surface of the spent BOC, Bi(012) dominates and on the surface of the spent BOI, both facets can be observed. From these results, it is inferred that Bi(003) and Bi(012) facets contribute differently to the electrochemical CO2 conversion reaction, with Bi(003) resulting in superior formic acid production. Considering their catalytic performance in terms of activity and selectivity (Fig. 2), we conclude that in the studied in situ-activated Bi catalyst system, the in situ-formed Bi(003) facet is more catalytically active than Bi(012). Furthermore, we conclude that bromide promotes exposure of the active Bi(003) surface sites, whereas chloride results in preferred exposure of a less-active Bi(012) surface. Iodide has a less profound influence on the facet exposure, resulting in equal contribution of Bi(003) and Bi(012) and intermediate performance for formic acid production.

a–c, Potential-dependent in situ XRD patterns of BOB (a), BOI (b) and BOC (c) in CO2-saturated 0.1 M KHCO3 solution, plotted in arbitrary units (a.u.) versus diffraction angle 2θ. d–f, Diffraction intensity of Bi(003) and Bi(012) facets as a function of applied potential of BOB (d), BOI (e) and BOC (f) in CO2-saturated 0.1 M KHCO3 solution. The purple and green shaded areas give the expected diffraction angles for the Bi(003) and Bi(006) reflections and Bi(012) reflections, respectively. The inserted structural models show the Bi(003) and Bi(012) facets from the top view.
Halide-guided reconstruction rates through in situ XANES
To further investigate the dynamic electronic structure of the studied BiOX catalysts during catalysis, in situ XANES was used under time-dependent and potential-dependent conditions (time resolution 2 min per spectrum). The measurement spot in the in situ XANES measurement was 1 × 1 µm and this was moved around during catalysis in order to minimize possible beam-damage effects. In Fig. 5a, BOB shows a typical BiOBr XANES spectrum at the OCP. When a cathodic bias of −1.15 V versus RHE is applied, the Bi L3-edge gradually moves to the lower energy region and the shape of the XANES curve starts changing, indicating restructuring and a change in oxidation state. As a function of time, the Bi L3-edge moves close to the edge energy of metallic Bi (13,419 eV), suggesting that BOB is being gradually converted into the metallic Bi phase under the reaction conditions. A similar trend is observed for BOI and BOC (Supplementary Figs. 34–36), in which the Bi L3-edge also gradually shifts to lower energy towards the edge position of metallic Bi. Next, we turn to the observation of BiOX reconstruction towards metallic Bi during catalysis. The rate of the structure transformation varies depending on the halide. The difference in rate of reconstruction can be seen more easily in the heat maps of the first derivative of the XANES spectra. It can be seen that the structure evolution has an earlier onset in BOI (4 min, Supplementary Fig. 34) and BOB (4 min, Fig. 5b), compared to BOC (8 min, Supplementary Fig. 35). This is in line with the in situ XRD and Raman spectroscopy measurements discussed above. In addition, the transition state (that is the mixture of BiOX and metallic Bi phase) of structure reconstruction persists longer in BOB, in contrast to BOI and BOC in which the transition state disappears faster (Fig. 5c).

a,b, Time-dependent in situ XANES Bi L3-edge spectra (a) and the heat map of the corresponding first derivative (b) in BOB, measured at −1.15 V versus RHE and plotted as normalized intensity (in arbitrary units, a.u.) versus energy. c, LCF results of time-dependent in situ XANES spectra for BOB, BOI and BOC. d,e, Potential-dependent in situ XANES Bi L3-edge spectra (d) and the heat map of the corresponding first derivative (e) in BOB, measured from the OCP to −1.35 V versus RHE. f, LCF results of potential-dependent in situ XANES spectra for BOB, BOI and BOC. The light red rectangles mark the transition state during activation.
Through linear combination fitting (LCF), the dynamic composition change during catalysis can be better quantified. Supplementary Fig. 37 shows the LCF example of BOB in 12 min. In Fig. 5c, the LCF analysis was applied to the time-dependent in situ XANES spectra, with Bi powder and BiOX as reference materials. It becomes evident that the structural evolution (BiOBr < 90% and Bi > 10%) has an earlier onset in BOI and BOB than in BOC at an applied potential of –1.15 V versus RHE, that is 4 min, 4 min and 8 min, respectively. The difference in onset time compared to the in situ XRD and Raman spectroscopy measurements could be due to the difference in their detection accuracy and signal-to-noise ratio, whereas the overall trend is unaffected by the choice of characterization technique. The transition state is defined as the intermediate state between the two stable reference states: pristine Bi oxyhalide and metallic Bi. The transition state is determined to last for 14 min in BOB, while full transformation from pristine Bi oxyhalide to metallic Bi is achieved in 6 min for BOI and BOC.
In the potential-dependent in situ XANES spectra, the same trend can be found: the onset potential for BOB and BOI is lower than for BOC. In Fig. 5d, no obvious change in the Bi L3-edge of BOB is observed upon changing the applied potential from the OCP to −0.35 V versus RHE, but the edge shifts to lower energy at −0.55 V versus RHE and onwards. Upon scanning the potential more negative, both the shape and edge position become similar to that of metallic Bi. Likewise, BOI has a structure evolution onset potential at −0.35 V versus RHE and quickly turns into a metallic-like structure at higher cathodic bias (Supplementary Fig. 38). On the contrary, BOC seems to be more difficult to be converted into the metallic counterpart, evidenced by the delayed reconstruction onset potential at −1.35 V versus RHE (Supplementary Fig. 39). In BOB, the transition state persists in a larger potential window, in contrast to that in BOI and BOC (Fig. 5e and Supplementary Figs. 38–40). The different onset (time and potential) of the BiOX reconstruction could be attributed to their different thickness and size: the larger and thicker BOC needs more driving force to initiate restructuring than BOB and BOI. However, after the reconstruction onset, the lifetime of the transition state seems to play an important role in tuning the Bi facet exposure during catalysis, which is guided by the presence of the halide.
The LCF results of the potential-dependent in situ XANES spectra are shown in Fig. 5f. The onset potential of structure evolution in BOB is identified to be −0.55 V versus RHE and the transition state is found in the range −0.55 to −0.95 V versus RHE. In BOI, the potential window where the transition state could be discerned ranges from −0.35 to −0.75 V versus RHE, while in BOC the restructuring occurs at −1.35 V versus RHE. In addition to the different size of the pristine BiOX, the onset of structure evolution during catalysis can also be explained by the hard–soft–acid–base theory, which states that soft acids form stronger bonds with soft bases, whereas hard acids form stronger bonds with hard bases43. Bi3+ is an intermediate Lewis acid, which tends to form a stronger bond with the intermediate Lewis base Cl– compared to the soft bases Br– and I–, in line with the Raman spectroscopy results. Therefore, more energy is needed to break Bi–Cl bonds and to further form metallic Bi. Furthermore, when the Bi–Cl bonds are finally broken and Bi monomers are formed, a sudden burst of nucleation and growth of metallic Bi occurs, evidenced by the short-lived transition state in the in situ XANES results, causing the preferred formation of edge planes (Bi(012) in this case over ordered basal planes, for example Bi(003)). The long-lived transition state indicates that BOB and BOI could be continuously reduced into metallic Bi, which is inferred to be beneficial for the formation of ordered basal plane structures with optimized catalytic performance for CO2 conversion into formic acid33.
Conclusions
Layered {001}-oriented Bi oxyhalides (that is BiOX, with X = Cl, Br or I) electrocatalysts were synthesized and used as templates for in situ formation of Bi electrocatalysts in the selective CO2 reduction reaction to formic acid. Using a GDE flow cell, the partial current density of formic acid in the prepared BiOBr (BOB) catalyst could reach up to 148 mA cm–2 with a near unity formic acid selectivity at −1.05 V versus RHE. Furthermore, formic acid selectivities of 76 and 69% were observed for BOI and BOC at −1.08 V versus RHE and −1.09 V versus RHE, at current densities of 95 and 88 mA cm–2, respectively. Through in situ Raman spectroscopy and XRD, it was found that the pristine BiOX electrocatalysts are converted into metallic Bi, but that the formation of the active sites is heavily influenced by the halide: bromide promotes Bi(003), chloride results in preferred Bi(012) exposure and iodide has an equal contribution of Bi(003) and Bi(012). This result combined with the catalytic performance trends suggests a structure–sensitivity relationship, in which the Bi(003) facet is more selective and active towards formic acid formation relative to the Bi(012) surface. In situ XAS measurements showed that the reconstruction of BOC proceeds rapidly, but at a higher cathodic bias, as compared to BOB and BOI. This suggests that the reconstruction rate of BiOX could determine facet exposure during catalysis in the presence of halides. This work provides insights into the active site of in situ-activated Bi-based electrocatalysts in the eCO2RR through multiscale in situ characterization and potentially paves the way for the rational design of other electrocatalysts for renewable production of chemicals and fuels.
Methods
Catalyst preparation
The studied BiOX materials (with X = Br, Cl or I) were synthesized by hydrothermal synthesis, modified from previously reported work57,58,59. In the procedure to obtain BiOBr, 5 mmol Bi(NO3)3·5H2O (Sigma-Aldrich, reagent grade, 98.0%) and 5 mmol cetyltrimethylammonium bromide (Sigma-Aldrich, ≥98.0%) were dissolved in 100 ml ultrapure water at room temperature for 40 min with stirring, followed by the addition of 1 M NaOH (Sigma-Aldrich, reagent grade, ≥98.0%) solution to adjust the pH to 7. After 1 h stirring, 80% of the mixed solution was poured into a 100 ml Teflon-lined stainless steel autoclave. The autoclave was then heated to 170 °C for 17 h before being cooled to room temperature in air. The sample was centrifuged and washed three times with ethanol and ultrapure water. Finally, the powders obtained were dried in the air at room temperature. Similarly, 4 mmol Bi(NO3)3·5H2O and 4 mmol KCl (Sigma-Aldrich, BioXtra, ≥99.0%) were added to 60 ml ultrapure water at room temperature with continuous stirring in a typical procedure for preparing BiOCl. After stirring for 1 h, the suspension was poured into a 100 ml Teflon-lined stainless steel autoclave. The autoclave was heated to 160 °C for 24 h. After cooling, the precipitates were centrifuged and washed with ethanol and ultrapure water three times. Finally, the powders were air dried at room temperature. For the synthesis of BiOI, 1.5 mmol Bi(NO3)3·5H2O was dissolved in a solution of 20 ml ethanol (Sigma-Aldrich, ACS reagent, ≥99.5%), with stirring for 30 min to form solution A. KI (1.5 mmol) (Sigma-Aldrich, ACS reagent, >99.0%) was dissolved in 40 ml ultrapure water to form solution B. Solutions A and B were mixed together under vigorous stirring. Then, the mixed solution was transferred to a 100 ml Teflon-lined stainless steel autoclave. The autoclave was heated at 80 °C for 3 h. After cooling down, the resulting precipitates were collected by centrifugation and washed with ethanol and ultrapure water three times. Finally, the sample was dried in an oven at 60 °C for 12 h before further characterization.
Characterization
The phase structure of the studied materials was determined by XRD measurements on a Bruker D8 Phaser diffractometer equipped with a Cu Kα source (λ = 1.54056 Å) for both ex situ and in situ measurements. Grazing incidence X-ray diffraction (GIXRD) measurements were also conducted on the Bruker D8 Phaser diffractometer using a θ of 0.3°. The morphology was studied using a scanning electron microscope (Phenom ProX). AFM measurements were performed with a Bruker Multimode microscope instrument, using silicon NSC−16 SCANASYST−AIR (F = 0.4 N m−1, fres = 70 kHz) in ScanAsyst mode. The local crystal structure, morphology and elemental distribution within the particles were determined by conducting HAADF-STEM and STEM-EDS measurements on an aberration-corrected Thermo Fisher Scientific Titan electron microscope operated at 300 kV and equipped with the ChemiSTEM system60.
Electrochemical measurements
An Ivium Compactstat.h10800 potentiostat was used for electrochemical performance measurements. In a gas-tight H-cell with two separated chambers, a standard three-electrode system was built with a proton exchange membrane (Nafion 117, Dupont). An Ag/AgCl electrode (ET069−1, 5 mm) and a Pt-mesh were employed as the reference electrode and counter electrode, respectively. Before starting the measurements, 0.1 M KHCO3 (Sigma-Aldrich, ACS reagent, 99.7%) aqueous solution was saturated with CO2 for at least 30 min to obtain an electrolyte with pH 6.8. During the measurements, CO2 was continuously delivered into the cathodic chamber at a constant rate of 10 ml min−1. In a GDE flow cell, the same type of membrane was used. An Ag/AgCl electrode (LF−1, 1 mm) and a platinized Ti electrode were employed as the reference electrode and counter electrode, respectively. KHCO3 solution (1 M) was used as the electrolyte in the GDE flow cell. To prepare working electrodes, 5 mg catalyst powder was mixed with 20 µl 5 wt% Nafion (Alfa Aesar, Nafion D−520 dispersion) solution and 500 µl methanol (Sigma-Aldrich, ≥99.9%) to form the catalyst ink after ultrasonication for 2 h. The working electrode was fabricated by drop-casting the catalyst ink onto carbon paper. The ECSA was evaluated by the double-layer capacitance (Cdl) through the CV measurements at different scan rates in the non-Faradaic region. The solution resistance (Rs) was automatically determined by electrochemical impedance spectroscopy with a frequency range of 10 Hz to 100 kHz for the iR correction in 0.1 M KHCO3 electrolyte solution in the H-cell, whereas manual iR correction was performed for the GDE cell with 1 M KHCO3 electrolyte solution (Rs = 0.8 Ω). The measured potential values were converted to the RHE using equation (1), in which the potential is corrected for the iR-drop and the influence of the electrolyte pH.
The potential-dependent catalytic performance was probed using a potentiostatic (chronoamperometry) test at different cathodic potentials. Each potential was held for around 40 min.
Product analysis
Online gas chromatography, together with a thermal conductivity detector and a flame ionization detector, was used for analysis of gas products. For determination of liquid products, 1H NMR spectroscopy was employed using water suppression mode. After catalysis, 0.5 ml of electrolyte was extracted and mixed with 0.1 ml of deuterated water (Sigma-Aldrich, ≤1 ppm) as the lock solvent. Dimethyl sulfoxide (0.05 µl) (Sigma-Aldrich, ACS reagent, ≥99.9%) was added as the internal reference.
The Faradaic efficiency (FE) of gas products was calculated using equation (2)
The production rate of all products was calculated using equation (3)
where n represents the number of electrons involved to produce the related products from CO2 or H2O (for example 2 for CO and H2 and 12 for C2H4), F is the Faraday constant (96,485 C mol−1), c is the concentration of the product measured by gas chromatography, f is the gas flow rate (ml min−1), I is the average measured current in 1 min (A), Vm is the volume of 1 mol gas at the reaction temperature and pressure (24,451 ml mol−1) and S represents the geometric area of the electrode (cm2).
The FE of liquid products was calculated by using equation (4)
where n is the number of electrons transferred to form the desired product (for example 2 for HCOOH), F is the Faraday constant (96,485 C mol −1), M is the molar concentration of the liquid product, V is the liquid volume, I is the average measured current in 1 min (A) and t is the duration time (s).
In situ Raman spectroscopy measurements
A Renishaw inVia Raman microscope and a 532 nm excitation laser coupled with a Nikon N40X - NIR water-dipping objective were used for the in situ Raman spectroscopy measurements. The laser power was set to below 1.5 mW in order to avoid laser damage. The time interval for each spectrum was 10 s for all measured samples. The in situ Raman spectroscopy measurements were coupled with chronoamperometry measurements, for which an Autolab PGSTAT 101 potentiostat was used with a scanning rate of 50 mV s−1 in CO2-saturated 0.1 M KHCO3 electrolyte solution (pH 6.8), at a potential of −1.15 V versus RHE. The same in situ Raman cell as previously published by our group was used for these measurements (Supplementary Fig. 41)9.
In situ XRD measurements
A Bruker D8 Phaser diffractometer was employed for the in situ XRD measurements. In a custom-made in situ cell (Supplementary Fig. 42), a glassy carbon wafer (SIGRADUR K films, diameter 22 mm, thickness 180 µm) coated with catalyst, a Pt wafer and an Ag/AgCl electrode were used as the working electrode, counter electrode and reference electrode, respectively. A back-illumination configuration was used for the measurement under Bragg mode. The electrolyte (CO2-saturated 0.1 M KHCO3 solution) was introduced into the cell with a flow rate of 10 ml min−1. An Ivium Compactstat.h10800 potentiostat was used for the chronoamperometry tests in the time- and potential-dependent in situ XRD measurements.
In situ XANES measurements
The Bi L3-edge high-energy resolution fluorescence detection X-ray absorption near-edge spectroscopy (HERFD-XANES) measurements were performed at the high-brilliance beamline ID26 of the European Synchrotron Radiation Facility (ESRF) in Grenoble, France. The storage ring operated in uniform multi-bunch mode with an electron current of 200 mA. Three undulators (u35) generated the incoming radiation, which was monochromated using a cryogenically cooled Si(111) double crystal monochromator. The X-rays were focused by two bent Pd mirrors to a size of ~210 × 105 μm2 (horizontal × vertical) at the sample position. The detector was a Si Avalanche Photo-Diode (APD) with 200 μm thickness and 10 × 10 mm2 active area. The size of an X-ray focus spot on the sample surface was 1 µm × 1 µm. For each spectrum, the acquisition time was set to 2 min. The same home-made in situ cell that was used for in situ XRD measurements with back-illumination configuration was used again for the in situ XANES measurements. However, the cell was tilted 90° to meet the measurement condition (fluorescence mode, Supplementary Fig. 42). A glassy carbon wafer (SIGRADUR K films, diameter 22 mm, thickness 180 µm) coated with catalyst, a Pt wafer and an Ag/AgCl electrode were used as the working electrode, counter electrode and reference electrode, respectively. The electrolyte (CO2-saturated 0.1 M KHCO3 solution) was introduced into the cell with a flow rate of 10 ml min−1. An Ivium Compactstat.h10800 potentiostat was used for the chronoamperometry tests in the time- and potential-dependent in situ XAS measurements. Mass-flow controllers (Bronkhorst) controlled the gas atmosphere in the reactor. The preliminary data analysis and normalization were performed using the Data Analysis X-ray spectroscopy (DAXS) program available at the ESRF. The Athena software from the Demeter package was used for the LCF.
Data availability
The data that support the findings of this study are presented in the text and Supplementary Information. Source data are provided with this paper. Additional data and information are available from the corresponding authors upon request.
References
Chu, S. & Majumdar, A. Opportunities and challenges for a sustainable energy future. Nature 488, 294–303 (2012).
Ross, M. B. et al. Designing materials for electrochemical carbon dioxide recycling. Nat. Catal. 2, 648–658 (2019).
Renfrew, S. E., Starr, D. E. & Strasser, P. Electrochemical approaches toward CO2 capture and concentration. ACS Catal. 10, 13058–13074 (2020).
Birdja, Y. Y. et al. Advances and challenges in understanding the electrocatalytic conversion of carbon dioxide to fuels. Nat. Energy 4, 732–745 (2019).
Gao, D., Arán-Ais, R. M., Jeon, H. S. & Roldan Cuenya, B. Rational catalyst and electrolyte design for CO2 electroreduction towards multicarbon products. Nat. Catal. 2, 198–210 (2019).
Wagner, A., Sahm, C. D. & Reisner, E. Towards molecular understanding of local chemical environment effects in electro- and photocatalytic CO2 reduction. Nat. Catal. 3, 775–786 (2020).
de Ruiter, J. et al. Probing the dynamics of low-overpotential CO2-to-CO activation on copper electrodes with time-resolved Raman spectroscopy. J. Am. Chem. Soc. 144, 15047–15058 (2022).
Wu, L. et al. Stabilization effects in binary colloidal Cu and Ag nanoparticle electrodes under electrochemical CO2 reduction conditions. Nanoscale 13, 4835–4844 (2021).
An, H. et al. Sub-second time-resolved surface-enhanced Raman spectroscopy reveals dynamic CO intermediates during electrochemical CO2 reduction on copper. Angew. Chem. Int. Ed. 60, 16576–16584 (2021).
Nitopi, S. et al. Progress and perspectives of electrochemical CO2 reduction on copper in aqueous electrolyte. Chem. Rev. 119, 7610–7672 (2019).
Jin, S., Hao, Z., Zhang, K., Yan, Z. & Chen, J. Advances and challenges for the electrochemical reduction of CO2 to CO: from fundamentals to industrialization. Angew. Chem. Int. Ed. 133, 20795–20816 (2021).
Verma, S., Kim, B., Jhong, H. R. M., Ma, S. & Kenis, P. J. A. A gross-margin model for defining technoeconomic benchmarks in the electroreduction of CO2. ChemSusChem 9, 1972–1979 (2016).
Bushuyev, O. S. et al. What should we make with CO2 and how can we make it? Joule 2, 825–832 (2018).
Wesselmark, M., Lagergren, C. & Lindbergh, G. Methanol and formic acid oxidation in zinc electrowinning under process conditions. J. Appl. Electrochem. 38, 17–24 (2008).
Koh, J. H. et al. Facile CO2 electro-reduction to formic acid via oxygen bidentate intermediate stabilized by high-index planes of Bi dendrite catalyst. ACS Catal. 7, 5071–5077 (2017).
Huttunen-Saarivirta, E., Kuokkala, V. T., Kokkonen, J. & Paajanen, H. Corrosion effects of runway de-icing chemicals on aircraft alloys and coatings. Mater. Chem. Phys. 126, 138–151 (2011).
Enthaler, S., von Langermann, J. & Schmidt, T. Carbon dioxide and formic acid—the couple for environmental-friendly hydrogen storage? Energy Environ. Sci. 3, 1207–1217 (2010).
Spurgeon, J. M. & Kumar, B. A comparative technoeconomic analysis of pathways for commercial electrochemical CO2 reduction to liquid products. Energy Environ. Sci. 11, 1536–1551 (2018).
Lee, C. W., Cho, N. H., Nam, K. T., Hwang, Y. J. & Min, B. K. Cyclic two-step electrolysis for stable electrochemical conversion of carbon dioxide to formate. Nat. Commun. 10, 3919 (2019).
Li, Q. et al. Tuning Sn-catalysis for electrochemical reduction of CO2 to CO via the core/shell Cu/SnO2 structure. J. Am. Chem. Soc. 139, 4290–4293 (2017).
Wang, S., Guan, B. Y. & Lou, X. W. D. Construction of ZnIn2S4–In2O3 hierarchical tubular heterostructures for efficient CO2 photoreduction. J. Am. Chem. Soc. 140, 5037–5040 (2018).
Zhao, C. X., Bu, Y. F., Gao, W. & Jiang, Q. CO2 reduction mechanism on the Pb(111) surface: effect of solvent and cations. J. Phys. Chem. C 121, 19767–19773 (2017).
Yang, S. et al. Waste-derived copper-lead electrocatalysts for CO2 reduction. ChemCatChem 14, e202200754 (2022).
Fan, K. et al. Curved surface boosts electrochemical CO2 reduction to formate via bismuth nanotubes in a wide potential window. ACS Catal. 10, 358–364 (2020).
Guan, Y., Liu, M., Rao, X., Liu, Y. & Zhang, J. Electrochemical reduction of carbon dioxide (CO2): bismuth-based electrocatalysts. J. Mater. Chem. A 9, 13770–13803 (2021).
Xia, D. et al. Recent progress of Bi-based electrocatalysts for electrocatalytic CO2 reduction. Nanoscale 14, 7957–7973 (2022).
Mohan, R. Green bismuth. Nat. Chem. 2, 336 (2010).
Yao, D. et al. The controllable reconstruction of Bi-MOFs for electrochemical CO2 reduction through electrolyte and potential mediation. Angew. Chem. Int. Ed. 60, 18178–18184 (2021).
Yang, Z., Oropeza, F. E. & Zhang, K. H. L. P-block metal-based (Sn, In, Bi, Pb) electrocatalysts for selective reduction of CO2 to formate. APL Mater. 8, 060901 (2020).
Pander, J. E., Baruch, M. F. & Bocarsly, A. B. Probing the mechanism of aqueous CO2 reduction on post-transition-metal electrodes using ATR-IR spectroelectrochemistry. ACS Catal. 6, 7824–7833 (2016).
Walker, R. J. et al. Surface termination and CO2 adsorption onto bismuth pyrochlore oxides. Chem. Mater. 28, 90–96 (2016).
Zhang, W. et al. Liquid-phase exfoliated ultrathin Bi nanosheets: uncovering the origins of enhanced electrocatalytic CO2 reduction on two-dimensional metal nanostructure. Nano Energy 53, 808–816 (2018).
Wang, D. et al. Unravelling the electrocatalytic activity of bismuth nanosheets towards carbon dioxide reduction: edge plane versus basal plane. Appl. Catal. B 299, 120693 (2021).
Walsh, A., Payne, D. J., Egdell, R. G. & Watson, G. W. Stereochemistry of post-transition metal oxides: revision of the classical lone pair model. Chem. Soc. Rev. 40, 4455–4463 (2011).
Su, P. et al. Ultrathin bismuth nanosheets as a highly efficient CO2 reduction electrocatalyst. ChemSusChem 11, 848–853 (2018).
Han, N. et al. Ultrathin bismuth nanosheets from in situ topotactic transformation for selective electrocatalytic CO2 reduction to formate. Nat. Commun. 9, 1320 (2018).
Yang, H. et al. Selective CO2 reduction on 2D mesoporous Bi nanosheets. Adv. Energy Mater. 8, 1801536 (2018).
García de Arquer, F. P. et al. 2D metal oxyhalide-derived catalysts for efficient CO2 electroreduction. Adv. Mater. 30, 1802858 (2018).
Wang, L. et al. Promoting photoreduction properties via synergetic utilization between plasmonic effect and highly active facet of BiOCl. Nano Energy 57, 398–404 (2019).
Yang, S. et al. Near-unity electrochemical CO2 to CO conversion over Sn-doped copper oxide nanoparticles. ACS Catal. 12, 15146–15156 (2022).
Handoko, A. D., Wei, F., Jenndy, Yeo, B. S. & Seh, Z. W. Understanding heterogeneous electrocatalytic carbon dioxide reduction through operando techniques. Nat. Catal. 1, 922–934 (2018).
Zheng, Y. et al. Atomic interface engineering and electric-field effect in ultrathin Bi2MoO6 nanosheets for superior lithium ion storage. Adv. Mater. 29, 1700396 (2017).
Parr, R. G. & Pearson, R. G. Absolute hardness: companion parameter to absolute electronegativity. J. Am. Chem. Soc. 105, 7512–7516 (1983).
Riedinger, A. et al. An intrinsic growth instability in isotropic materials leads to quasi-two-dimensional nanoplatelets. Nat. Mater. 16, 743–748 (2017).
Marcandalli, G., Monteiro, M. C. O., Goyal, A. & Koper, M. T. M. Electrolyte effects on CO2 electrochemical reduction to CO. Acc. Chem. Res. 55, 1900–1911 (2022).
Zheng, W. iR compensation for electrocatalysis studies: considerations and recommendations. ACS Energy Lett. 8, 1952–1958 (2023).
Yang, K., Kas, R., Smith, W. A. & Burdyny, T. Role of the carbon-based gas diffusion layer on flooding in a gas diffusion electrode cell for electrochemical CO2 reduction. ACS Energy Lett. 6, 33–40 (2021).
Yang, K. D. et al. Morphology‐directed selective production of ethylene or ethane from CO2 on a Cu mesopore electrode. Angew. Chem. Int. Ed. 129, 814–818 (2017).
Yoon, Y., Yan, B. & Surendranath, Y. Suppressing ion transfer enables versatile measurements of electrochemical surface area for intrinsic activity comparisons. J. Am. Chem. Soc. 140, 2397–2400 (2018).
Zhang, D., Li, J., Wang, Q. & Wu, Q. High {001} facets dominated BioBr lamellas: facile hydrolysis preparation and selective visible-light photocatalytic activity. J. Mater. Chem. A 1, 8622–8629 (2013).
Jiang, M. et al. Anti-aggregation and morphology-controlled effects of bacterial cellulose encapsulated BiOBr for enhanced photodegradation efficiency. Cellulose 27, 8843–8858 (2020).
Wu, D. et al. Boron doped BiOBr nanosheets with enhanced photocatalytic inactivation of Escherichia coli. Appl. Catal. B 192, 35–45 (2016).
Denisov, V. N. et al. Raman spectra of superconducting Bi–Sr–Ca–Cu–O single crystals. Solid State Commun. 70, 885–888 (1989).
Kazyrevich, M. E. et al. Photocurrent switching effect on platelet-like BiOI electrodes: influence of redox system, light wavelength and thermal treatment. Electrochim. Acta 190, 612–619 (2016).
Weng, S., Pei, Z., Zheng, Z., Hu, J. & Liu, P. Exciton-free, nonsensitized degradation of 2-naphthol by facet-dependent BiOCl under visible light: novel evidence of surface-state photocatalysis. ACS Appl. Mater. Interfaces 5, 12380–12386 (2013).
Panagiotopoulou, P., Christodoulakis, A., Kondarides, D. I. & Boghosian, S. Particle size effects on the reducibility of titanium dioxide and its relation to the water–gas shift activity of Pt/TiO2 catalysts. J. Catal. 240, 114–125 (2006).
Feng, H. et al. Modulation of photocatalytic properties by strain in 2D BiOBr nanosheets. ACS Appl. Mater. Interfaces 7, 27592–27596 (2015).
Wang, L. et al. Monolayer epitaxial heterostructures for selective visible-light-driven photocatalytic NO oxidation. Adv. Funct. Mater. 29, 1–8 (2019).
Shao, D., Zhang, L., Sun, S. & Wang, W. Oxygen reduction reaction for generating H2O2 through a piezo-catalytic process over bismuth oxychloride. ChemSusChem 11, 527–531 (2018).
Schlossmacher, P., Klenov, D. O., Freitag, B. & von Harrach, H. S. Enhanced detection sensitivity with a new windowless XEDS system for AEM based on silicon drift detector technology. Micros. Today 18, 14–20 (2010).
Acknowledgements
B.M.W. acknowledges support from the Strategic UU–TU/e Alliance project ‘Joint Centre for Chemergy Research’ as well as from the Netherlands Center for Multiscale Catalytic Energy Conversion (MCEC), an NWO gravitation programme funded by the Ministry of Education, Culture and Science of the government of the Netherlands. S.B. acknowledges support from the European Research Council (ERC Consolidator Grant #815128 REALNANO). S.A. and T.A. acknowledge funding from the University of Antwerp Research fund (BOF). We also thank J. Wijten, J. Janssens and T. Prins (all from the Inorganic Chemistry and Catalysis group, Utrecht University) for helpful technical support. S. Deelen (Faculty of Science, Utrecht University) and L. Wu (Inorganic Chemistry and Catalysis group, Utrecht University) are acknowledged for the design of the in situ XRD cell. We also acknowledge B. Detlefs, P. Glatzel and V. Paidi (ESRF) for the support during the HERFD-XANES measurements on the ID26 beamline of the ESRF.
Author information
Authors and Affiliations
Contributions
B.M.W. and W.v.d.S conceived the idea and supervised the project. S.Y. synthesized the materials, performed the catalytic reactions, conducted the ex situ and in situ X-ray characterizations and analysed the data. S.A., S.B. and T.A. performed and supervised the electron microscopy measurements. H.A. and J.d.R. performed the in situ Raman measurements and analysed the data. H.W. performed the AFM measurements and assisted with the in situ XANES measurements, together with X.Y. The initial manuscript was written by S.Y. together with B.M.W. and W.v.d.S. All authors have given input on the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Catalysis thanks Arnav Malkani and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary Information
Supplementary Figs. 1–42 and Refs. 1 and 2.
Source data
Source Data Fig. 1
Statistical data used to plot the figures.
Source Data Fig. 2
Statistical data used to plot the figures.
Source Data Fig. 3
Statistical data used to plot the figures.
Source Data Fig. 4
Statistical data used to plot the figures.
Source Data Fig. 5
Statistical data used to plot the figures.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Yang, S., An, H., Arnouts, S. et al. Halide-guided active site exposure in bismuth electrocatalysts for selective CO2 conversion into formic acid. Nat Catal 6, 796–806 (2023). https://doi.org/10.1038/s41929-023-01008-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41929-023-01008-0
This article is cited by
-
In situ reconstruction of Bi nanoparticles confined within 3D nanoporous Cu to boost CO2 electroreduction
Science China Materials (2024)
