

Abstract
High-performance rechargeable batteries are becoming very important for high-end technologies with their ever increasing application areas. Hence, improving the performance of such batteries has become the main bottleneck to transferring high-end technologies to end users. In this study, we propose an argon intercalation strategy to enhance battery performance via engineering the interlayer spacing of honeycomb structures such as graphite, a common electrode material in lithium-ion batteries (LIBs). Herein, we systematically investigated the LIB performance of graphite and hexagonal boron nitride (h-BN) when argon atoms were sent into between their layers by using first-principles density-functional-theory calculations. Our results showed enhanced lithium binding for graphite and h-BN structures when argon atoms were intercalated. The increased interlayer space doubles the gravimetric lithium capacity for graphite, while the volumetric capacity also increased by around 20% even though the volume was also increased. The ab initio molecular dynamics simulations indicate the thermal stability of such graphite structures against any structural transformation and Li release. The nudged-elastic-band calculations showed that the migration energy barriers were drastically lowered, which promises fast charging capability for batteries containing graphite electrodes. Although a similar level of battery promise was not achieved for h-BN material, its enhanced battery capabilities by argon intercalation also support that the argon intercalation strategy can be a viable route to enhance such honeycomb battery electrodes.
Export citation and abstract BibTeX RIS
1. Introduction
For the last couple of decades, energy storage materials have become paramount due to the high demand for indispensable electronic devices, from mobile appliances to electric vehicles. Technological expectations from a novel energy storage unit are higher power supply and/or energy density, longer cycle life, and shorter charging time than its predecessors. Today, the best option that gradually meets these expectations is lithium-ion batteries (LIBs) [1, 2]. LIBs basically consist of positive and negative electrodes called cathode and anode parts, in which the materials are used mainly to determine the electrochemical performance. Beside lithium alloyed metals, graphite and carbon-based materials are still widespread anode materials used in commercial LIBs due to their abundance, relatively low-cost, and high electronic conductivity [3]. Graphite is an attractive anode material due to graphene's unique electronic structure and high thermal conductivity. Due to the graphene's high surface area as a 2D crystal, it can accommodate more lithium ions [4, 5]. Besides the many excellent features of the graphene, its lithium intercalation capacity is limited (specific capacity is 372 mAh g−1 for LiC6 [6]), which may not satisfy the ever increasing demand of future technologies. Several methods are reported in the literature to address this issue: One approach is to find alternative materials such as metal oxides, phosphorous, silicon, and composite structures [7–14]. However, although higher specific capacities are reached for several materials, poor cycling and rate performances are still a drawback [15].
Moreover, lower electrical conductivity compared to graphite and significant volume changes leading to damage in the battery are still challenging. One another approach is the applications of various operations on the graphene material itself. For example, the defects created in graphene have caused an increase in the binding energy and diffusion rate of lithium [16, 17]. Alternatively, graphene structure can also be doped with various elements [18], and/or functional groups can be added between graphene layers [19, 20] which also leads to an increase in a specific capacity. Nevertheless, the desired level of specific capacity and rate performances has yet to be achieved.
Recently, hexagonal boron nitride (h-BN), which has an analogous structure to graphene, has also started to be considered in recent studies for battery applications. Besides the structural similarities, as the atomic size of boron and nitrogen are similar to carbon, h-BN has low electrical conductivity because of its wide band gap [21]. Nonetheless, Zhang et al reported that functionalized h-BN nanosheets have high specific capacity (up to 400 mAh g−1) and good cycle stability [22]. Chowdhury et al showed that h-BN, used as a capping agent for black-phosphorene, enhances the binding energy and reduces the lithium diffusion energy barrier [21]. Hosseini et al indicated that the functionalized BN—fullerene structure could be used as an anode, and this structure has higher cell voltage than carbon nanotubes and C24 fullerene [23]. Although a lot of work has been done on using the h-BN structure as an anode material [24], its use at the moment is not very promising, as the battery performance is lower than that of graphene [25]. Still doping graphene by h-BN [18] or heterostructuring them [15], may increase the carrier mobility and widen the bandgap. Therefore, h-BN has been started to investigate as a partner of graphene in new generation electronics to improve battery performance.
In light of the literature above, high battery performance means higher storage capacities, ion mobilities, and charging/discharging rates. In this study, we pursued a strategy to improve the battery performance of graphite and h-BN by the intercalation of argon between their layers. Our previous experimental studies inspired this idea. The graphite and h-BN particles were added to modify the thermal conductivity and mechanical properties of SiAlON and SiC matrix phases, respectively [26, 27]. In these studies, while SiAlON/graphite samples were produced by spark plasma sintering (SPS) of the mixture of SiAlON-forming powders (Si3N4, AlN, Al2O3, CaCO3, Sm2O3) and micro-fluidized graphite flakes at 1950 °C under 50 MPa for 10 min, SiC/h-BN composite samples were produced by SPS of SiC and h-BN forming powders (boric acid and urea) at 1850 °C under 50 MPa for 17 min [28]. SPS'ed SiAlON/graphite and SiC/h-BN ceramic composite samples were cut into 3 mm discs before mechanically thinning down to 30 µm then, these samples were bombarded with Ar+ ions from two oppositely sided ion guns operated at 6 keV energy and 8° angle; with parallel surfaces of the samples by using Bal-TEC RES101 ion beam milling equipment. Ion implantation geometry is summarized in figure 1(a). In the same figure, scanning transmission electron microscope (Jeol 2100 F) analyses show that the ceramic composites contain either h-BN (figure 1(b)) or graphite (figure 1(c)) particles. The energy dispersive x-ray spectroscopy (EDX) chemical analysis spectra confirm that additive h-BN and graphite particles contain argon (Ar) element (figures 1(d) and (e)), while matrix particles do not. According to these studies, Ar+ ions can be experimentally implanted between h-BN and graphite basal planes. Based on quantitative EDX analysis results, average Ar content was found as between 0.5%–2.1% atomic ratio in h-BN and graphite samples. However, variation in the Ar content results from relative orientation difference between (0002) basal planes of h-BN and graphite with respect to the Ar ion gun direction. It is worth noting that Ar+ ion implantation (intercalation) can only occur if the specific orientation condition between basal planes of h-BN or graphite and Ar+ source has been satisfied according to the electron energy loss spectroscopy (EELS) & EDX results and details can be accessed from [26].

Figure 1. (a) Ion beam implantation schematics, scanning transmission electron microscope bright field (STEM-BF) images of (b) h-BN, (c) graphite containing ceramic composites and EDX spectra taken from Ar+ implanted (d) h-BN and (e) graphite particles. Asterisks (*) mark indicate the point where EDX spectra were taken. (Please note that the Si-K peak in the spectra resulted from the internal fluorescence peak of the EDX detector.)
Download figure:
Standard image High-resolution imageThe scope of this work is to examine how battery performance changes when an Ar-intercalated h-BN or graphite is used as an anode material in LIBs, which was aimed to reduce the diffusion energy barrier of lithium and increase its specific capacity. In this regard, first-principles based density functional theory (DFT) calculations were performed to investigate the stabilities, lithium binding energies, lithium diffusion energy barriers, and lithium storage capacities of Ar-intercalated graphite and h-BN nanosheets.
2. Computational details
All DFT calculations were performed by first-principles code Vienna ab initio simulation package [29, 30]. The electron-ion interactions were treated by the projector-augmented wave method [31]. The generalized gradient approximation within the Perdew–Burke–Ernzerhof was used to treat the exchange-correlation potential [32]. The plane-wave energy cut-off was set to 500 eV. As a model system, 5 × 5 supercell structures (on XY plane) of graphite and h-BN were chosen as it is firstly a large enough size to overcome periodic interactions of single adsorbent Ar and/or Li, and secondly, it is an affordable size within our computational limits to simulate a low Ar atomic ratio by 1.96% to enable large space for alkali ions such as Li. Furthermore, the chosen 5 × 5 supercell model is also commensurate with the experimental EDX analyses in terms of the Ar atomic ratio. After the Ar-intercalation to the electrode materials, the unit cell vectors are kept frozen. At the same time, all the atomic coordinates are optimized to see whether Li atoms could fit in the elaborated space without compromising energetic stability. The Brillouin-zone integration was performed within the Monkhorst−Pack scheme by using a regular Γ centered 2 × 2 × 2 k-points mesh [33]. The convergence criteria for electronic and ionic relaxations were set to 10−6 eV and 10−2 eV Å−1, respectively. Van der Waals correction of Grimme (D3) method with Becke–Johnson damping was employed to model the interaction at the interface [34, 35]. The migration energy barriers for argon and lithium atoms among graphite and h-BN layers were calculated using the nudged elastic band (NEB) method [36] by considering the pathway between two adjacent lowest energy binding sites. To quantify the charge distribution between Li atoms and h-BN or graphite structures, the Bader charge analysis [37, 38] was used. The molecular dynamic simulations (MD) [39, 40] were performed within the isobaric-isothermal (NPT) ensemble [41, 42]. MD simulations were performed as each simulation lasting 8 ps with a time step of 1 fs. Only 300 K was considered the simulation temperature, assuming the temperature does not change during charge/discharge.
3. Ar intercalated graphite and h-BN systems
Firstly, we constructed and optimized the bulk and double-layer graphite and h-BN supercell structures. AB stacking type unit cell for graphite [43] and AA stacking type for h-BN [44, 45] were created, which are the most stable stacking types, respectively. Afterward, the possible sites of the argon atoms in the structure were investigated systematically by also considering the relative positions of Ar atoms between adjacent layers.
The Ar binding energy (Eb[Ar]) was calculated as follows:
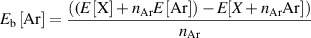
where E[X] is the total energy of graphite or h-BN system. E[![$X + {n_{{\text{Ar}}}}{\text{Ar]}}$](https://content.cld.iop.org/journals/0953-8984/35/8/085301/revision3/cmaca8e7ieqn1.gif) is the total energy of the Ar intercalated system.
is the total energy of the Ar intercalated system.  is the number of Argon atoms, and
is the number of Argon atoms, and ![$E\left[ {{\text{Ar}}} \right]$](https://content.cld.iop.org/journals/0953-8984/35/8/085301/revision3/cmaca8e7ieqn3.gif) is the total energy of a single Ar atom in a box. According to this definition, positive binding energies define favorable binding, and negative binding energies can be considered an energetical cost. Initially, Ar binding to graphene and h-BN monolayers was calculated, and the hollow site was found to be the preferred binding site with less than 5 meV differences. The predicted Ar binding energies were 92 and 85 meV on graphene and h-BN, respectively. However, the binding energies become negative when Ar intercalated between two monolayers, and the calculated energy costs were 2.07 and 1.89 eV for graphene and h-BN double layers, respectively. This is because the Ar intercalation increases the interlayer separation by 3 Å for graphene and 2 Å for h-BN, and therefore the interaction between layers diminishes. Note that a hollow site relative to each layer is not possible for graphite due to its AB stacking. Still, in graphite, the hollow-atop site is more favorable than the atop–atop site relative to both layers by 10 meV in graphite. For h-BN, the hollow site is more favorable than both atop sites (atop of B or N) by 94 meV. All the considered Ar positions and their relative energies are given also in supplementary info (table S1). When Ar is intercalated in bulk graphite, we considered first where there is one Ar atom for every two layers. In this case, the interlayer separation increases only between Ar-containing layers and by a reduced amount of 2.3 Å. Still, the energetical cost increased to 2.63 eV compared to argon intercalated between two graphene monolayers.
is the total energy of a single Ar atom in a box. According to this definition, positive binding energies define favorable binding, and negative binding energies can be considered an energetical cost. Initially, Ar binding to graphene and h-BN monolayers was calculated, and the hollow site was found to be the preferred binding site with less than 5 meV differences. The predicted Ar binding energies were 92 and 85 meV on graphene and h-BN, respectively. However, the binding energies become negative when Ar intercalated between two monolayers, and the calculated energy costs were 2.07 and 1.89 eV for graphene and h-BN double layers, respectively. This is because the Ar intercalation increases the interlayer separation by 3 Å for graphene and 2 Å for h-BN, and therefore the interaction between layers diminishes. Note that a hollow site relative to each layer is not possible for graphite due to its AB stacking. Still, in graphite, the hollow-atop site is more favorable than the atop–atop site relative to both layers by 10 meV in graphite. For h-BN, the hollow site is more favorable than both atop sites (atop of B or N) by 94 meV. All the considered Ar positions and their relative energies are given also in supplementary info (table S1). When Ar is intercalated in bulk graphite, we considered first where there is one Ar atom for every two layers. In this case, the interlayer separation increases only between Ar-containing layers and by a reduced amount of 2.3 Å. Still, the energetical cost increased to 2.63 eV compared to argon intercalated between two graphene monolayers.
Then we constructed one argon atom per layer unit cells to imitate realistic systems by also considering the steric effect of one Ar atom on other Ar atom positions between adjacent layers. As the argon atoms were sent into the layers, two extremes would be the case for Ar atoms: either they will stay in line or try to avoid each other. Thus, two different cases (Z1 and Z2) have been considered depending on the relative positions of Ar atoms between consecutive layers. The case where the Ar atoms coincide on the same position in the XY-plane (atop) was defined as Z1 (figures 2(a) and (c)), while the other case where the Ar atoms are optimally apart from each other was described as Z2 (figures 2(b) and (d)). In graphite structures, argon atoms were on a hollow site relative to one layer while atop a site relative to the other one for Z1 and Z2 cases. For the Z1 condition of the h-BN structure, both argon atoms sit at the hollow sites relative to both layers, while for the Z2 case, the second Ar atom would coincide to atop positions relative to both layers to maximize the Ar–Ar separation.

Figure 2. The top and side views of relaxed graphite layers for Z1 and Z2 configurations are given in (a) and (b), respectively. For relaxed h-BN layers, same configurations are given in (c) and (d). Here, d is the interlayer separation; the argon atom was shown as a turquoise ball. The hallow and atop sites are also denoted in (d).
Download figure:
Standard image High-resolution imageThese two cases, Z1 and Z2, were specifically selected and constructed as they are extreme cases for the chosen 5 × 5 unit cell, where the Z1 case did not distort the layers sterically. In contrast, the Z2 case distorts the layers at maximum. There are several reasons for initially choosing this 5 × 5 supercell size model, besides experimental hints on argon content in h-BN and graphite (0.5%–2.1% atomic ratio). When argon atoms are inserted into graphite or h-BN layers, they replace potential lithium adsorption sites. Thus, a high Ar concentration is not desirable. Furthermore, a high concentration of Ar atoms may also sterically trap Li ions between layers and/or might cause significant distortions on C or h-BN layers. On the other hand, to model very low Ar concentrations, much larger supercell structures should be considered, which limits the practicality of the NEB and MD calculations at a quantum mechanical level. Moreover, too low concentrations of Ar will also behave like a defect besides a widening agent in the interlayer distance of layers. For this 5 × 5 supercell construction of graphite and h-BN systems, the Ar atomic ratio to C or BN was 1.96%.
Without Ar insertion, the optimized interlayer separation of graphite is 3.34 Å. When the argon atom is inserted between the layers, the separation distance between the layers is increased to 6.10 Å for Z1 and 4.95 Å for Z2 cases. Energetically, the Z2 case is slightly more favorable than the Z1 case by 22 meV for a 5 × 5 graphite unit cell. Similar to the graphite case, when argon atoms were inserted between layers of h-BN, the structural integrity was preserved throughout the layers in both cases. At the same time, there is a curling behavior of the layers around the intercalated Ar atoms in Z2 structures. The interlayer separation distance was increased to 5.71 Å and 4.34 Å for Z1 and Z2 cases, respectively, from the initial value of 3.29 Å in the pristine h-BN. The energetical difference between Z1 and Z2 cases was found to be 0.43 eV, and the Z2 case is energetically more favorable than the graphite case. The energetical preference of the Z2 case over Z1 means that the argon atoms try to spread on the XY-plane by avoiding each other on consecutive layers.
4. Binding of Li to Ar-intercalated systems
In this part of the study, the intercalation of lithium atoms between argon-intercalated h-BN and graphite layers was investigated. First, we analyzed how a single Li atom interacts with an Ar atom in free space to estimate how closely they can interact. There was a very weak interaction (∼30 meV) between these atoms with an optimum distance of around 3.88 Å. The repulsive interaction dominates for distances lower than 3 Å, as seen in figure 3.

Figure 3. Energy changes depending on the distance between lithium and argon atoms.
Download figure:
Standard image High-resolution imageThen, the binding energies were calculated for a single lithium atom inside the Ar-intercalated graphene and h-BN systems for both Z1 and Z2 structures. The Li binding energy (Eb[Li]) was calculated as follows:
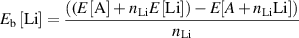
where E[A] is the total energy of graphite or h-BN system with intercalated argon atoms. E[![$A + {n_{{\text{Li}}}}{\text{Li]}}$](https://content.cld.iop.org/journals/0953-8984/35/8/085301/revision3/cmaca8e7ieqn4.gif) is the total energy of the same system, including Li atoms.
is the total energy of the same system, including Li atoms.  is the number of lithium atoms, and
is the number of lithium atoms, and ![$E\left[ {{\text{Li}}} \right]$](https://content.cld.iop.org/journals/0953-8984/35/8/085301/revision3/cmaca8e7ieqn6.gif) is the total energy of a single lithium atom in a box.
is the total energy of a single lithium atom in a box.
The lithium atom preferred the center of the hexagons (hollow site) by avoiding atop position of individual atoms on graphite and h-BN layers. Since graphite has AB stacking, even if a lithium atom is at a hollow position on one layer, it has to be in atop according to the other layer. However, if both interactions are atop according to carbon layers, the energetical cost was 0.27 eV. On the other hand, in h-BN, lithium atom can sit at hollow sites relative to both layers. In this case, the energetical cost for the atop interactions was found to be 0.14 eV. All the considered Li positions and their relative energies are given also in supplementary info (table S1).
With the intercalation of argon atoms into graphite and h-BN structures, the lithium binding energy increased in general for both Z1 and Z2 cases. For the Z1 case, the binding energy increased by 0.1 eV for graphene while 0.16 eV for h-BN, and for the Z2 case, it increased by 0.5 eV for graphene and 0.25 eV for h-BN. Interestingly, the intercalation of argon widened the distance between the layers to accommodate more Li atoms and enhanced the Li binding energy to the system. The reason for the increased binding energy could be the reduced van der Waals interactions between the diverging layers from each other due to the Ar intercalation. Lithium's binding energy is very low on h-BN, so their electrodes are not common in the literature. However, lithium binding energy can be improved with Ar intercalation. This strategy may be possible for their usage in battery applications if the binding energies stay favorable for higher coverages.
Higher lithium coverage systems, including monolayer and double layers of lithium, were constructed to check the battery capacities of graphite and h-BN and to see whether they can be increased by Ar intercalation. The binding energy change of such lithium coverages was given in figure 4 with respect to the number of adsorbed lithium atoms on a 5 × 5 × 1 unit cell for both pristine and argon intercalated (Z2 case) graphite and h-BN systems. Without Ar intercalation, full coverage Li monolayer (50 Li atoms for a 5 × 5 cell) is the limit as the binding energy diminishes with the adsorption of the second monolayer of lithium on top, and the structural integrity deteriorates. However, Li binding energy stays favorable for the Ar-intercalated graphite system even when a second full layer of lithium is inserted. As seen in figure 4, the binding energy of lithium in the graphite structure is almost linear from 15 to 65 lithium, and there is a slight decrease until reaching double full coverage. Hence, the specific capacity of graphite can be at least doubled by Ar-intercalation.

Figure 4. The calculated binding energy versus the number of intercalated Li atoms for pristine and Ar-intercalated graphene and h-BN systems. X (purple) marks indicate deterioration in structural integrity. Here for 5 × 5 surface supercell systems, 25 Li atoms can be treated as a full layer coverage. In an inset double layer, lithium coverage in Ar-intercalated graphite, the system was shown where the blue sphere denotes argon, green spheres represent lithium, and the brown sphere denotes carbon atoms.
Download figure:
Standard image High-resolution imageIn the case of Ar-intercalated h-BN, the structure readily deteriorated when adding a few lithium atoms after the full monolayer of lithium coverage. On the other hand, the Ar-intercalation increased the Li capacity considerably for the h-BN system up to lithium monolayer coverage by increasing the lithium binding energy from 0.36 eV to 1.13 eV. When compared to the graphite case, the average Li binding energies of h-BN systems are lower than graphite with and without Ar intercalation, but still, the energy differences were lowered by the Ar intercalation for these systems. This is particularly important for h-BN as it cannot solely be used as electrode material in its pristine form due to the low Li binding energies. With this, the counterintuitive average Li binding energy increase from single Li to monolayer for h-BN can be partly attributed to the Li-Li interactions.
5. Battery performance of Ar-intercalated systems
To obtain information about the electrochemical properties of the argon intercalated graphite and h-BN structures, we estimated the open-circuit-voltage (OCV) values by calculating averaged cell voltages over a range of lithium-ion concentrations (x) by using common half-cell reactions. As the common half-cell reaction can be written as:
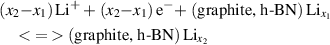
for the charge/discharge process for graphite or h-BN systems, the OCV values can then be calculated by the formula:
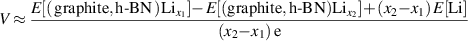
where ![$E[\,({\text{graphite, h-BN}}){\text{L}}{{\text{i}}_{{x_1}}}\,]$](https://content.cld.iop.org/journals/0953-8984/35/8/085301/revision3/cmaca8e7ieqn7.gif) and
and ![$E[\,({\text{graphite, h-BN}}){\text{L}}{{\text{i}}_{{{\text{x}}_{\text{2}}}}}\,]$](https://content.cld.iop.org/journals/0953-8984/35/8/085301/revision3/cmaca8e7ieqn8.gif) are the total energies of graphite or h-BN systems with x1 and x2 lithium atom intercalated, respectively. E[Li] is the total energy of the alkali metal.
are the total energies of graphite or h-BN systems with x1 and x2 lithium atom intercalated, respectively. E[Li] is the total energy of the alkali metal.
The OCV value measures the battery performance, and positive voltage values are necessary for the considered concentrations to be achieved. Figure 5 shows calculated OCV profiles of Ar-intercalated graphite and h-BN systems. Looking at the voltage profiles, one can see that all the values are positive even up to the second full layer intercalation of lithium. However, it should be noted that the structural integrity has deteriorated in our calculations for the h-BN system after the first lithium monolayer. On the other hand, OCV values were generally higher than h-BN and over 1.25 V up to almost 1.2 layer coverage for the Ar-intercalated graphite system. Afterward, it drops to levels around 0.50 and 0.75 V for higher coverages between 1.2–1.5 and 1.5–2.0, respectively. In general, the OCV profile shows a linear behavior until monolayer coverage. It then starts to drop step by step, although there is a possible artificial increase beyond 1.5 layer coverage due to the selected adsorption sites. Indeed, to understand the OCV profile more precisely in terms of accurate voltage values, all the possible configurations for each lithium concentration should be investigated, which is beyond our computational limits and the scope of this study. Despite that, a smooth voltage profile for an Ar-intercalated graphite system up to high coverages is very promising for battery applications, and the help of Ar-intercalation can achieve high lithium capacities. Nevertheless, the increasing voltage values for the h-BN system can be explained by the increased average binding energy for higher Li coverages, which is partly attributed to the Li–Li interactions as the Li binding to the h-BN usually is very low. In addition, a sharp decline in the voltage values is because of the unstability of the high coverage h-BN systems.

Figure 5. Calculated open circuit voltage, V, profile for Li intercalation as a function of Li concentration (x). Concentration 1 is defined as full coverage of the structure.
Download figure:
Standard image High-resolution imageGravimetric and volumetric specific capacities are also significant performance criteria for battery applications that need to be addressed. Gravimetric capacity (GC) (mAh g−1) is basically the energy that can be provided in ampere-hours by one gram electrode material and can be calculated theoretically by the formula:
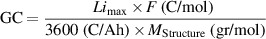
where  is the maximum number Li concentration that can be intercalated,
is the maximum number Li concentration that can be intercalated,  is Faraday constant, and
is Faraday constant, and  is the molecular weight of the electrode. As seen in table 1, GC was over doubled for the Ar-intercalated graphite as it accommodated the second layer of lithium between its layers with the help of the increased interlayer spacing. However, for the h-BN case, the GC did not improve as the extra layer of lithium is not possible and even decreases slightly because the Ar atom intercalated occupies one of the possible sites for Li already. Although the Ar atoms also occupied some Li sites (2 sites out of 50 possible in a 5 × 5 supercell) in the Ar-intercalated graphite case, this is negligible as the possibility of an extra lithium layer (50 more sites) dominates the capacity. Although the GC doubled for the graphite case, the increase is related to the increasing interlayer space; hence the volume of Ar-intercalated graphite is larger. Thus, we also compared the volumetric capacities to ensure the improvement by Ar intercalation. Different from the GC, volumetric capacity (VC) (mAh cm−3) is basically the energy that can be provided per volume of electrode material and can be calculated by the formula:
is the molecular weight of the electrode. As seen in table 1, GC was over doubled for the Ar-intercalated graphite as it accommodated the second layer of lithium between its layers with the help of the increased interlayer spacing. However, for the h-BN case, the GC did not improve as the extra layer of lithium is not possible and even decreases slightly because the Ar atom intercalated occupies one of the possible sites for Li already. Although the Ar atoms also occupied some Li sites (2 sites out of 50 possible in a 5 × 5 supercell) in the Ar-intercalated graphite case, this is negligible as the possibility of an extra lithium layer (50 more sites) dominates the capacity. Although the GC doubled for the graphite case, the increase is related to the increasing interlayer space; hence the volume of Ar-intercalated graphite is larger. Thus, we also compared the volumetric capacities to ensure the improvement by Ar intercalation. Different from the GC, volumetric capacity (VC) (mAh cm−3) is basically the energy that can be provided per volume of electrode material and can be calculated by the formula:
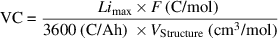
Table 1. The list of calculated gravimetric and volumetric capacity according to the number of intercalated lithium atoms.
| Structure | System | Gravimetric capacity (mAh g−1) | Volumetric capacity (mAh cm−3) |
|---|---|---|---|
| Pristine graphite | 50 C + 50 Li | 1115.7 | 2528.3 |
| Ar-intercalated graphite | 50 C + 2 Ar + 98 Li | 2050.4 | 3352.6 |
| Pristine h-BN | 25 B + 25 N + 50 Li | 1079.9 | 2482.9 |
| Ar-intercalated h-BN | 25 B + 25 N + 2 Ar + 49 Li | 994.3 | 1864.9 |
where  is the molecular volume of the total system. The VC of pristine graphite was calculated as 2.53 Ah cm−3 by considering the full coverage of Li, and it dropped to 1.64 Ah cm−3 for the Ar-intercalated case. However, as the layer separation allowed more Li accommodation in the Ar-intercalated case, the VC value increased to 3.35 Ah cm−3 when a double layer of Li is considered, which is 32% larger than pristine graphite.
is the molecular volume of the total system. The VC of pristine graphite was calculated as 2.53 Ah cm−3 by considering the full coverage of Li, and it dropped to 1.64 Ah cm−3 for the Ar-intercalated case. However, as the layer separation allowed more Li accommodation in the Ar-intercalated case, the VC value increased to 3.35 Ah cm−3 when a double layer of Li is considered, which is 32% larger than pristine graphite.
The thermal stability of electrodes is also very crucial in battery applications for safety and long-term usage. Therefore, the thermal stability of graphite and h-BN electrodes with full Li loading was examined by an NPT ensemble of ab initio MD simulations. MD simulations were performed for 8 ps at 300 K for 5 × 5 supercell structures to check whether any structural transformation or distortion would happen at room temperature. Figure 6 shows the Ar-intercalated graphite system's total energy and temperature variations during MD simulations. Thermal and energy fluctuations show that the equilibrium is reached quite rapidly for the considered MD parameters. During the 8 ps simulation time (8000-time step), the adsorbed Li atoms did not agglomerate and stayed on their respective adsorption sites on graphite layers (see figure 6 inset crystal structures). Furthermore, the fluctuations of graphene layers were at expected levels, and no structural transformation was observed, confirming the thermal stability of this system at room temperature. Hence, it can be thought that this will preserve the stability of a realistic battery for a longer period of time which in turn would result in an increase in battery life.

Figure 6. Temperature and total energy evaluation profiles in MD simulations for full coverage of Li-ion intercalation in argon-intercalated graphite. Corresponding crystal structures were given as insets at 0, 2, 4, 6, and 8 ps.
Download figure:
Standard image High-resolution imageLi mobility on the electrode material is another critical parameter for battery performance, and low Li diffusion barriers are desired for high charge/discharge rates. NEB calculations were performed to determine the migration energy barriers (selected between two adjacent lowest energy binding sites) for both argon and lithium atoms among graphite and h-BN layers.
Firstly, the lithium diffusion barriers of h-BN and graphite electrodes with and without Ar intercalation have been calculated and compared to each other. The diffusion path was selected between two adjacent lowest energy binding sites for Li on a 5 × 5 cell, as shown in figure 7. Both sites are center of hexagons according to the bottom layer and equivalent to each other without Ar intercalation, while the energies differ slightly (less than 2 meV) with Ar-intercalation as the symmetry was broken. Without Ar-intercalation, the Li diffusion energy barrier in h-BN (0.426 eV) is much larger than graphite (0.140 eV), which makes pristine h-BN, not a plausible electrode material. When we compare the Li migration energy on graphite with literature, our value of 140 meV is significantly smaller than the usually reported values around 0.4–0.5 eV for graphite [46–48]. However, all these values are obtained for LiC6 crystals, which have AA stacking of graphite.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Figure 7. Diffusion barrier profiles and optimized migration pathways between two nearest lowest energy sites of Li atom. (a) is for 5 × 5 graphite and (b) is for h-BN.
Download figure:
Standard image High-resolution image{kind=link}
On the other hand, our result is quite similar to Leggesse et al (0.117 eV) [49], which uses the same AB stacking within a 3 × 3 graphite supercell. With Ar intercalation, both barriers are lowered drastically and surprisingly, the barrier of Li in h-BN becomes utilizable levels, which is lower than pristine graphite (∼0.116 eV). Consistently, this diffusion energy barrier value for Ar intercalated h-BN is also very similar to the Li migration barrier on a monolayer h-BN (0.10 eV) [50]. For Ar intercalated graphite system, the drop of the Li diffusion energy barrier is highest (16 meV) when the Li atom is relatively far from the Ar atom, although still there is quite a significant drop (63 meV) in the diffusion energy barrier when the Li atom moves closely around the intercalated Ar atom. Almost one order decreasing from 140 to 16 meV with the help of Ar-intercalation for graphite hints more than 100 times higher mobility at room temperature with a crude approximation from the Arrhenius law (D ≈ exp(−Ea/kb T)). The reason for the accelerated diffusion in the Ar-intercalated system can be attributed to the weakened interactions between layers such as van der Waals. The same level of Li diffusivity in Ar-intercalated h-BN, compared to graphite, could promote h-BN as a candidate electrode material; however, its storage capacity would not be doubled by the Ar-intercalation as in the graphite case.
Determining the diffusion energy barrier of argon is also essential for the stability of the system if it is meant to be used in battery applications. If argon's binding energy and diffusion to the system are lower than that of lithium, argon may move quickly away from the system. Therefore, the diffusion energy barrier of argon in the system is also calculated and compared to Li diffusion. Two different cases were considered for the Ar diffusion in the systems (see supplementary figure S1). In the first case, one of the argon atoms was kept fixed while the other was moving, which can also be viewed as a transformation between the extreme Z1 and Z2 cases. The energy barrier for this case was found to be 0.056 eV, which is considerably larger than for Li migration barrier in graphite. In the second case, we examined the diffusion of a third argon atom in the denser Z2 structure. The energy barrier for this case was found to be 0.031 eV, which is almost double for the Li case but lower than the first case. Altogether these considerably larger diffusion energy barriers of Ar atoms than Li atoms confirmed that the Li atoms are more mobile than the Ar atoms in the graphite electrode material.
When we compare the Li atom diffusion energy barriers with other suggested materials in the literature, we see the enhanced value for graphite via Ar-intercalation (16 meV) is considerably lower than commercially used anode materials based on TiO2 (0.35–0.55 eV)[51–53] and also lower than largely studied novel 2D materials such as MXenes (0.04 eV for Mo2C)[54], TMDs (0.25 eV for MoS2)[55], or black phosphorus (0.08 eV)[56].
As the last analysis for investigating the battery performance of these materials, the amount of charge transfer between Li atom and electrode systems was determined using the Bader charge analysis. According to the Bader charge analysis, Li atoms ionized almost perfectly (∼0.98 e−) for each system independent from the coverage as well (see table 2). The transferred charge from the Li atoms into the system spread over the layers of the graphite system. On the h-BN system, the transferred charge was taken from N atoms but spread overall N atoms in general. Surprisingly for single Li intercalation, the transferred charge also did not localize on the closest C or N atom.
Table 2. List of average charge per element according to the bader charge analysis for graphite and h-BN systems. The following columns also give the minimum and maximum values of the atomic charges corresponding to analyzed elemental groups.
| Structure | Element (number) | Average charge (e−) | Minimum charge (e−) | Maximum charge (e−) |
|---|---|---|---|---|
| Graphite + Li (single) | C (100) | −0.009 | 0.092 | 0.154 |
| Li (1) | 0.988 | |||
| Graphite + 50 Li (monolayer) | C (100) | −0.491 | −0.502 | −0.486 |
| Li (50) | 0.983 | |||
| Graphite + Ar | C (100) | −0.001 | −0.082 | 0.098 |
| Ar (2) | 0.026 | 0.025 | 0.026 | |
| Graphite + Ar + Li (single) | C (100) | −0.010 | −0.154 | 0.085 |
| Ar (2) | 0.026 | 0.024 | 0.025 | |
| Li (1) | 0.983 | |||
| Graphite + Ar + 48 Li (monolayer) | C (100) | −0.465 | −0.743 | −0.193 |
| Ar (2) | −0.432 | −0.432 | −0.431 | |
| Li (48) | 0.987 | 0.985 | 0.989 | |
| h-BN + Li (single) | B (50) | 2.969 | 2.967 | 2.972 |
| N (50) | −2.989 | −3.062 | −2.961 | |
| Li (1) | 0.989 | |||
| h-BN + 50 Li (monolayer) | B (50) | 2.967 | 2.966 | 2.968 |
| N (50) | −3.952 | −3.953 | −3.951 | |
| Li (50) | 0.984 | 0.984 | 0.984 | |
| h-BN + Ar | B (50) | 2.970 | 2.969 | 2.972 |
| N (50) | −2.972 | −3.045 | −2.931 | |
| Ar (2) | 0.036 | 0.035 | 0.037 | |
| h-BN + Ar + Li (single) | B (50) | 2.970 | 2.969 | 2.972 |
| N (50) | −2.992 | −3.134 | −2.996 | |
| Ar (2) | 0.032 | 0.031 | 0.032 | |
| Li (1) | 0.995 | |||
| h-BN + Ar + 49 Li (monolayer) | B (50) | 2.969 | 2.966 | 2.972 |
| N (50) | −3.867 | −4.771 | −3.366 | |
| Ar (2) | −0.214 | −0.201 | −0.228 | |
| Li (46) | 0.986 | 0.979 | 0.988 |
6. Conclusions
In summary, we demonstrated that the interlayer distance expansion by argon intercalation could be a promising strategy to increase h-BN and graphite battery performance. Compared to their corresponding pristine structures, argon-intercalated h-BN and graphite structures show higher Li mobilities, and graphite has a larger battery capacity.
Enlarged interlayer distance helps the intercalation of the second layer of Li ions between graphite layers, which increases the GC more than twice. Meanwhile, the VC was also increased by 20.3% for graphite. Although there is an improvement for the Ar-intercalated h-BN system as well in terms of Li adsorption energy and diffusion barriers, the storage capacity did not increase as much as in the case of graphite. Bader charge analysis showed that the Li atoms could donate their electrons to the system for all the considered coverages. The OCV profiles hinted that the calculated high lithium capacities could be attained. Furthermore, ab-initio MD simulations indicated that the fully loaded Ar-intercalated graphite system is thermally stable at around room temperature.
Hence, this study showed that argon atoms could be stably put into between graphite and h-BN layers, increasing the battery performance. Therefore, argon intercalation could serve as a general method to expand interlayer distance and might be utilized for other 2D materials as well. The increased interlayer space may also accommodate larger metal ions such as Na+ and multivalent ions such as Ca2+ and may be utilized in alternative battery applications to Li.
Acknowledgments
Computational resources were provided by the High Performance and Grid Computing Center (TRGrid e-Infrastructure) of TUBITAK ULAKBIM and the National Center for High Performance Computing (UHeM) of Istanbul Technical University. ID and CS acknowledge the support from Eskisehir Technical University (ESTU-BAP 22ADP150, 21GAP085 and 22ADP111).
Data availability statement
The data that support the findings of this study are available upon reasonable request from the authors.
Conflict of interest
There are no conflicts to declare.
Author contributions
The manuscript was written through the contributions of all authors. All authors have given approval for the final version of the manuscript.
Supplementary data (1.1 MB PDF)
Supplementary data (1.4 MB PDF)