

Abstract
Cold atmospheric pressure plasmas in and in contact with liquids represent a growing field of research for various applications. Understanding the interactions between the plasma generated species and the liquid is crucial. In this work we perform molecular dynamics (MD) simulations based on a quantum mechanical method, i.e. density-functional based tight-binding (DFTB), to examine the interactions of OH radicals and O atoms in bulk water. Our calculations reveal that the transport of OH radicals through water is not only governed by diffusion, but also by an equilibrium reaction of H-abstraction with water molecules. Furthermore, when two OH radicals encounter each other, they either form a stable cluster, or react, resulting in the formation of a new water molecule and an O atom. In addition, the O atoms form either oxywater (when in singlet configuration) or they remain stable in solution (when in triplet configuration), stressing the important role that O atoms can play in aqueous solution, and in contact with biomolecules. Our observations are in line with both experimental and ab initio results from the literature.
Export citation and abstract BibTeX RIS
The interaction of cold atmospheric pressure plasmas (CAPPs) with liquid surfaces has been gaining increasing interest over the past decades for various applications, like water treatment and material synthesis, as well as in plasma medicine [1–3]. In recent years many studies have been conducted to gain insight in the plasma–liquid interaction (see e.g. [4–9]). Recently, an extensive overview has been presented on plasma–liquid interactions [10] stating the upcoming challenges as well as the unresolved questions. Obviously, more fundamental studies are required, based on both experiments and computer simulations. Computational research, both on the macroscopic scale and the atomic scale, can be very useful to gain better insight in the underlying mechanisms [11–14]. Macroscopic investigations, such as fluid dynamics and 0D chemical kinetics modeling [12–14], provide a global representation of the experimental mechanisms, which allows easy comparison with experiments, but they require detailed information like reaction rate coefficients as input for the model. This detailed information can be provided by atomic scale simulations, such as molecular dynamics (MD).
Such MD simulations have been performed by Yusupov et al, investigating the behavior of various reactive oxygen species (ROS) in water [11]. It was observed that hydroxyl radicals (OH) and hydrogen peroxide molecules (H2O2) are able to penetrate the gas–liquid interface, quickly entering the liquid, and that O atoms and OH radicals penetrate the liquid interface through H-abstraction reactions, resulting in the formation of new OH radicals in the bulk liquid. A classical force field was used in the MD simulations to describe the time-dependent behavior of the ROS. Although such simulations describe the investigated system in a reactive and dynamic manner, the force field parametrization defines the accuracy of the results. Recent experimental work [4] showed that transport of O atoms into the liquid is an effective process, leading to an increasing concentration of O in water, which was not observed in the simulations. This led us to believe that the classical force field might not be accurate enough to describe the interactions of the O atoms with water, and that more accurate methods, based on quantum mechanics, might be needed. The latter are, however, computationally more intensive, limiting the time and length scales of the systems to be simulated. Recently, it was demonstrated that the density-functional based tight-binding (DFTB) method is more suitable to describe the behavior of oxygen-based radicals than a classical force field, as the electron distribution of each individual atom is taken into account so that radicals can be characterized (i.e. defining unpaired electrons) [15]. Indeed, using this technique we were able to successfully describe the interactions between OH radicals and the TM6 part of the membrane protein P-glycoprotein [16].
Therefore, in this work we apply the DFTB method to examine the interaction of oxygen-based radicals, i.e. OH and O, with water molecules. DFTB is a computational technique, which utilizes a Taylor series expansion of the Kohn–Sham total energy as used in DFT calculations [17]. We use here third order DFTB (i.e. DFTB3 complemented with the 3ob-2-1 parameter set), which employs the third series expansion of the mentioned energy equation, resulting in a self-consistent computational method. DFTB3 is able to accurately describe hydrogen binding energies, proton affinities and proton transfers, for systems up to 103 atoms [15–18]. Thus, it should provide a more realistic description of the interaction of oxygen-based radicals with water than classical force fields.
Figure 1 shows a snapshot of the molecular system investigated. A simulation box with dimensions of ±15 Å × 15 Å × 15 Å, containing water molecules, ensuring a density of 1 mg ml−1 around the introduced radicals, was equilibrated at room temperature in the canonical ensemble, employing the Nosé–Hoover thermostat [19]. Subsequently, 1 or 2 H2O molecules were replaced with either OH radicals or O atoms, and the interactions were simulated for 10 ps, using a time step of 0.25 fs. It should be noted that small dimensions are used in this investigation, resulting in relatively high concentrations. However, the aim of our work is to study the interactions between the radicals, dissolved in water, at the moment they take place. Here, the assumption is made that two radicals in experimental conditions will eventually meet each other and interact. Minimal effect of the simulation box on the results are expected as the introduced radicals are embedded in several shells of water molecules, greatly reducing the influence of long range interactions. In DFTB3 the number of unpaired electrons must be specified as an input parameter which remains constant throughout the whole simulation, i.e. 1 for each OH radical in the system, 0 for singlet O and 2 for triplet O.

Figure 1. Illustration of the investigated molecular system: a box of H2O molecules, where 1 H2O molecule is replaced by an O atom (black circle). H and O atoms are depicted in white and red, respectively.
Download figure:
Standard image High-resolution imageIntroducing a single OH radical in the water system resulted in a stable solution, as previously reported by Yusupov et al [11]. A significant affinity between the OH radical and H atoms of the surrounding water molecules was observed, yielding H-transfer, according to the following equilibrium reaction:
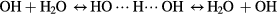
This observation suggests that the transport of OH radicals through the water is not only by diffusion, but also through reactions with water molecules, as also reported by Yusupov et al [11].
However, in the case of two OH radicals in the water system, the situation is more complex. Two different events were encountered during 10 identical simulation runs: (i) a H-abstraction between the two radicals, resulting in a single O atom and a water molecule (see figure 2), or (ii) a side-to-side complex of the two radicals (see figure 3). Both events were encountered 5 and 2 times, respectively. Interestingly, the formation of this side-to-side complex reduces the interactions with the surrounding water molecules, i.e. no H-transfer was observed after the formation of this complex. This is most likely caused by the O···O interaction which decreases the attractive forces towards the H-atoms of the surrounding water molecules. This is further supported by the fact that the observed complex remains intact over the remaining course of the simulation. The reason for the difference in number of observations between the two interactions can be attributed to two phenomena. First, the H-abstraction often occurred during an interaction of the second OH radical with water (as mentioned above). Second, the formation of the side-by-side complex requires a certain orientation of interaction between the two OH radicals and this lowers the probability for the formation of this complex, compared to the H-abstraction reaction. However, no statistical conclusion can be drawn given the limited number of interactions simulated.
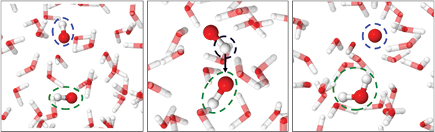
Figure 2. Snapshots of three consecutive iterations of the DFTB simulations, displaying the H-abstraction between the two introduced OH radicals (indicated by the bottom green and top blue dashed circle) (left figure). After the formation of a head-to-tail interaction between the two OH radicals (middle figure), a H-abstraction occurs (indicated by the arrow), resulting in the formation of atomic O and a water molecule (bottom green circle), stable in the aqueous solution (right figure). The chemical species of interest are depicted with solid spheres, while the surrounding water molecules are depicted as semitransparent sticks. O and H are shown in red and white, respectively.
Download figure:
Standard image High-resolution image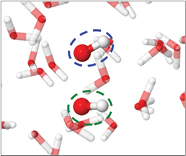
Figure 3. Snapshot of the side-to-side interaction of two OH radicals (indicated with the bottom green and top blue dashed circles). The chemical species of interest are depicted with solid spheres, while the surrounding water molecules are depicted as semitransparent sticks. O and H are shown in red and white, respectively.
Download figure:
Standard image High-resolution imageBoth observations are different from the previous classical MD simulations of Yusupov et al, where only 1 OH radical at a time was simulated, and only the first event was observed. They are, however, in line with ab initio calculations performed by Codorniu-Hernández in 2014 [20], reporting both the production of O atoms, when two OH radicals are found in a head-to-tail formation (figure 2), and the formation of a metastable side-to-side complex, like our observations in figure 3.
Interesting is the fact that the formation of H2O2 has not been observed during the simulations, as it is prevented by the presence of an energy barrier [20]. As such, longer time scales, beyond the reach of DFTB (which is situated in the picosecond range), are required for these reactions to take place. Furthermore, DFTB is unable to accurately describe this energy barrier. However, this has been estimated using ab initio calculations performed by Codorniu-Hernández [20].
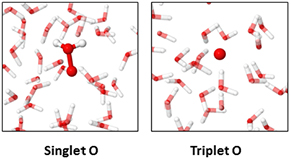
Because our calculations predict that the OH radicals give rise to O atom formation inside the water, we also investigated the behavior of O atoms in bulk water, for both singlet O (i.e. no unpaired electrons), and triplet O (i.e. two unpaired electrons). The results for both systems are depicted in figure 4. In the case of singlet O (figure 4 left), the formation of oxywater (O  OH2 complex) was observed in all simulations, even within the first iterations, and the observed complex remained stable in the solution throughout the rest of the simulation. Despite the fact that oxywater is the metastable isomer of hydrogen peroxide (with a free energy of formation of the latter of about −29 kcal mol−1 [20]), the formation of H2O2 is not observed during the simulated time scale, suggesting that this transition should have a (small) energy barrier, as also reported in literature [20].
OH2 complex) was observed in all simulations, even within the first iterations, and the observed complex remained stable in the solution throughout the rest of the simulation. Despite the fact that oxywater is the metastable isomer of hydrogen peroxide (with a free energy of formation of the latter of about −29 kcal mol−1 [20]), the formation of H2O2 is not observed during the simulated time scale, suggesting that this transition should have a (small) energy barrier, as also reported in literature [20].
{kind=link}
{kind=link}
{kind=link}
Figure 4. Snapshot of the interactions between an O atom and surrounding water molecules, for singlet O (left) and triplet O (right). When singlet O is introduced, the formation of oxywater is observed, while triplet O remains stable in solution. The chemical species of interest are depicted using solid spheres, while the surrounding water molecules are depicted as semitransparent sticks. O and H atoms are shown in red and white, respectively.
Download figure:
Standard image High-resolution image{kind=link}
However, in the case of triplet oxygen, no reaction was observed within the simulated time scale, indicating the stability of this species inside bulk water (see right figure 4). This correlates well with the formation of a stable triplet O atom after the head-to-tail interaction of two OH radicals, as depicted in figure 2. The stability of triplet O in the liquid is supported by the experimental work of Hefny et al [4], who observed both the oxidation of phenol and the formation of O2 when O atoms were introduced in the plasma system, and more interestingly, no increase in H2O2 was observed, indicating the absence of OH radicals in the treated solution. These observations thus clearly support the stability of triplet O atoms in water, as predicted by our DFTB simulations. Moreover, as no increase in H2O2 is observed experimentally, we expect that triplet O occurs more than singlet O (as oxywater) in solution, as also suggested by Codorniu-Hernández [20].
In summary, we used DFTB MD simulations to investigate the behavior of OH radicals and O atoms in bulk water. Our results indicate that the transport of OH radicals in bulk water is not only governed by diffusion, but also through an equilibrium reaction of H-abstraction with water molecules, in line with the work of Yusupov et al [11]. However, when an OH radical encounters another OH radical, some distinct reactions are observed. When both radicals interact through a head-to-tail interaction, a H-abstraction occurs, resulting in the formation of a new water molecule and a triplet O atom, which remains stable in the aqueous solution. On the other hand, when both OH radicals form a side-to-side configuration, a cluster, stable in solution, is observed. These observations are supported by ab initio calculations [20]. Finally, when investigating the behavior of both singlet and triplet O atoms in bulk water, our calculations reveal that singlet O interacts with water to create oxywater within the first few interactions of the MD simulations, which is expected to form H2O2, while triplet O remains stable throughout the simulated time scale of 10 ps. The latter is in line with the experimental work of Hefny et al [4]. This observation highlights the importance of atomic O in the chemistry of plasmas in contact with liquids. Indeed, given the high reactivity of atomic O with RONS and biomolecules, we expect that this species will play a crucial role in the processes found in the upper layers of the treated solution and cannot be neglected when investigating the further mechanisms of RONS in water and with biomolecules.
Acknowledgments
The authors thank Peter Bruggeman (University of Minnesota, USA) and Jan Benedikt (Ruhr-Universität Bochum, Germany) for the interesting discussions regarding the existence of O in aqueous solutions. Furthermore, they acknowledge financial support from the Fund for Scientific Research (FWO) Flanders (project number G012413N). The calculations were performed using the Turing HPC infrastructure at the CalcUA core facility of the Universiteit Antwerpen, a division of the Flemish Supercomputer Center VSC, funded by the Hercules Foundation, the Flemish Government (department EWI) and the Universiteit Antwerpen.