Within-Generation Polygenic Selection Shapes Fitness-Related Traits across Environments in Juvenile Sea Bream

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
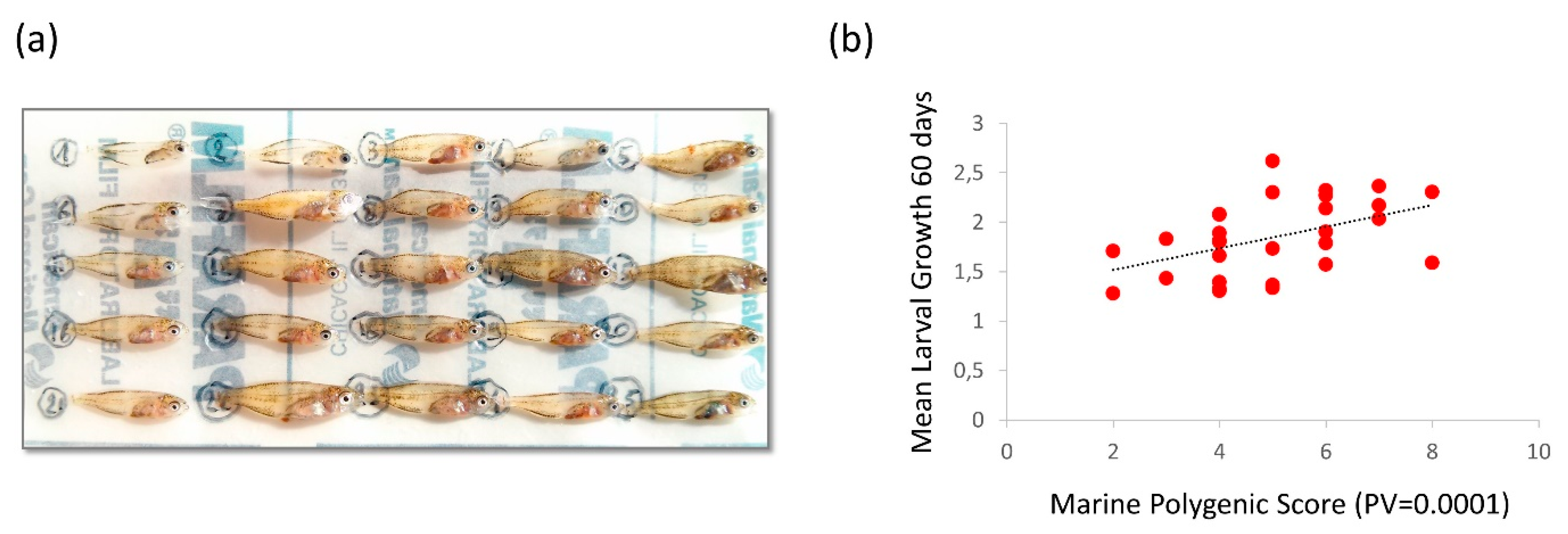{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sampling
2.2. Scoring of Phenotypes
2.3. RAD Sequencing, Variant Calling and Individual Genotyping
2.4. Genetic Homogeneity among Samples
2.5. Test for Single-Generation Selection
2.6. Estimating the Survival Probability of Genotypes
2.7. Genotype–Phenotype Links
3. Results
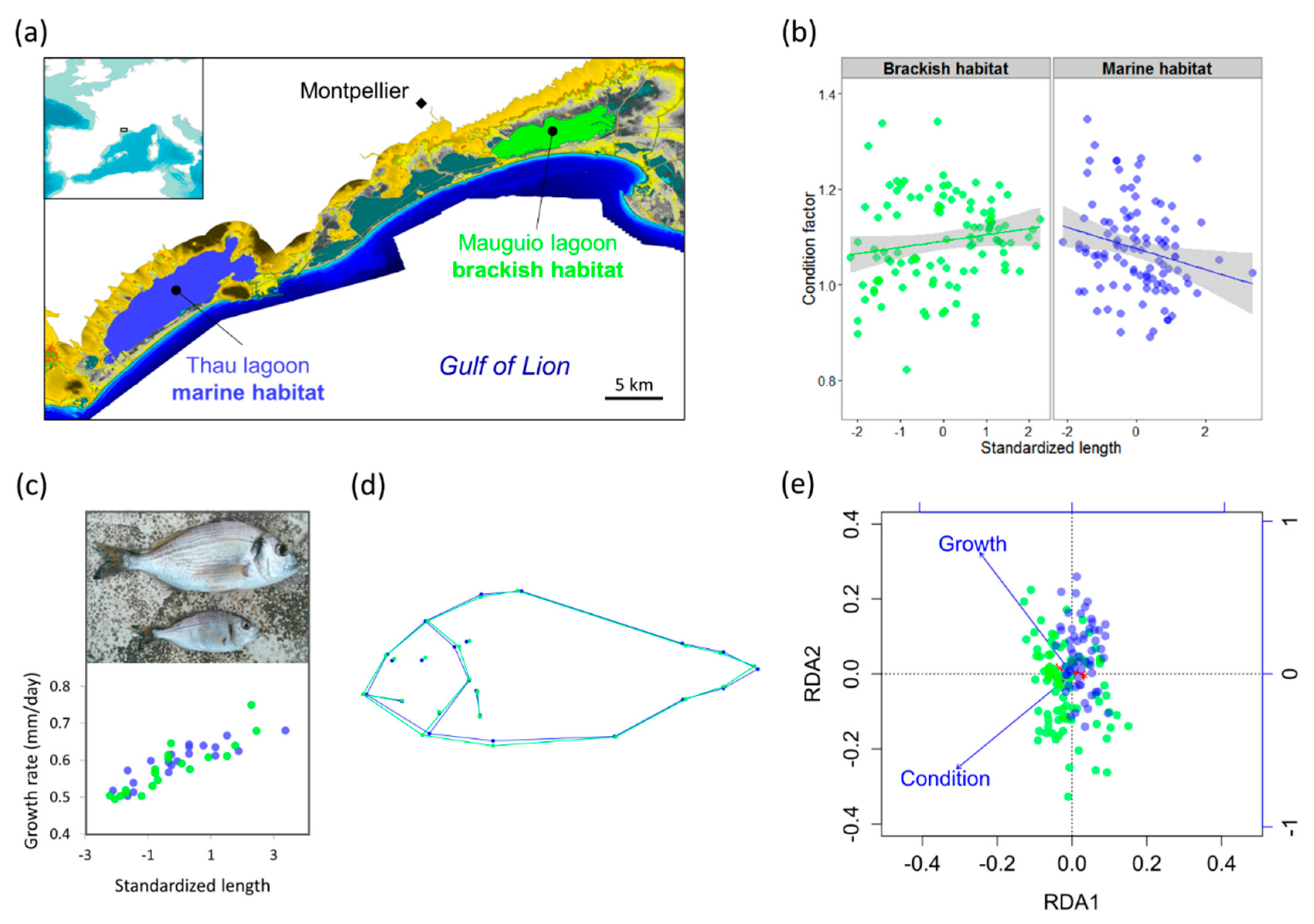
3.1. Phenotypic Variation
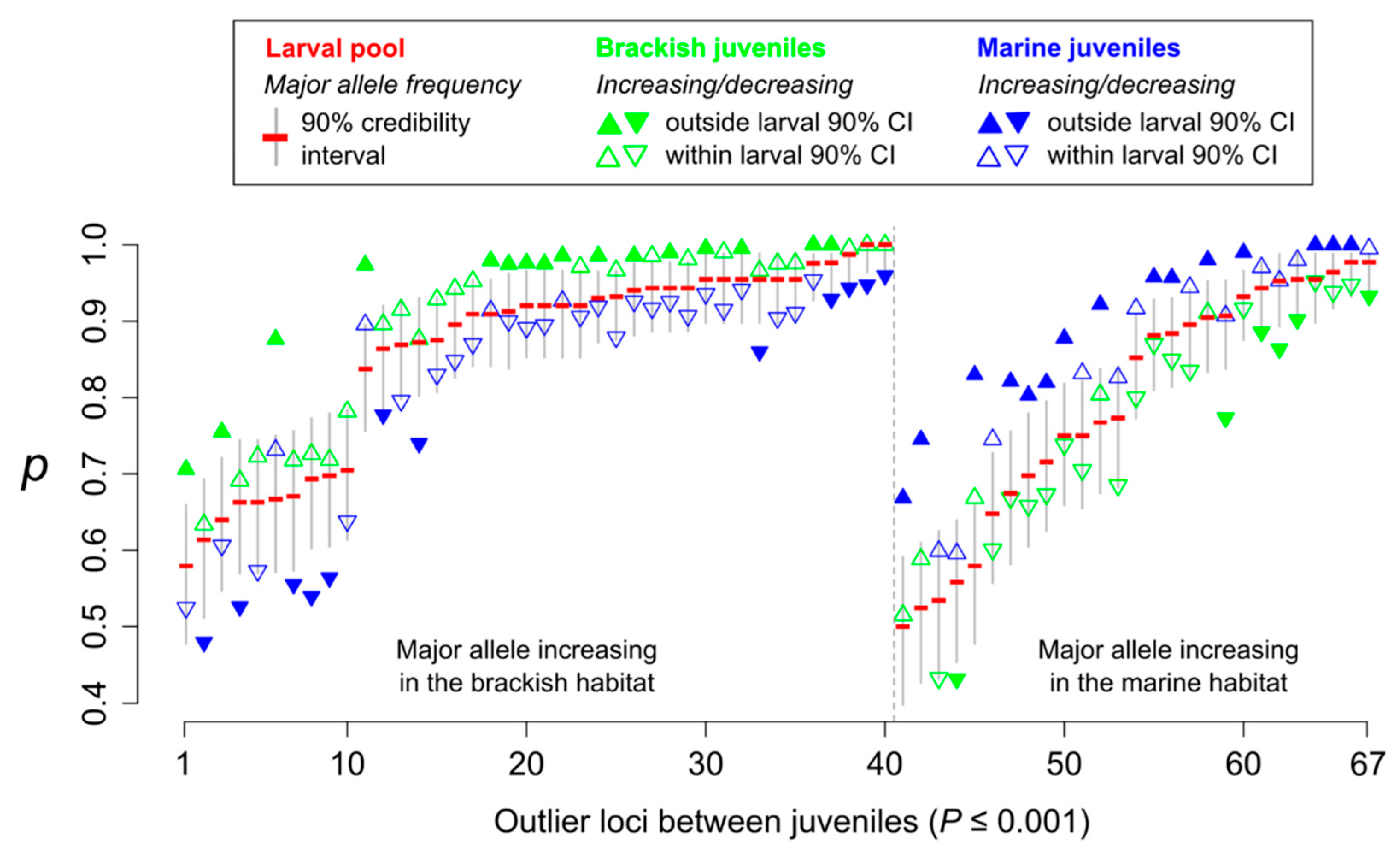
3.2. Population Genetic Homogeneity
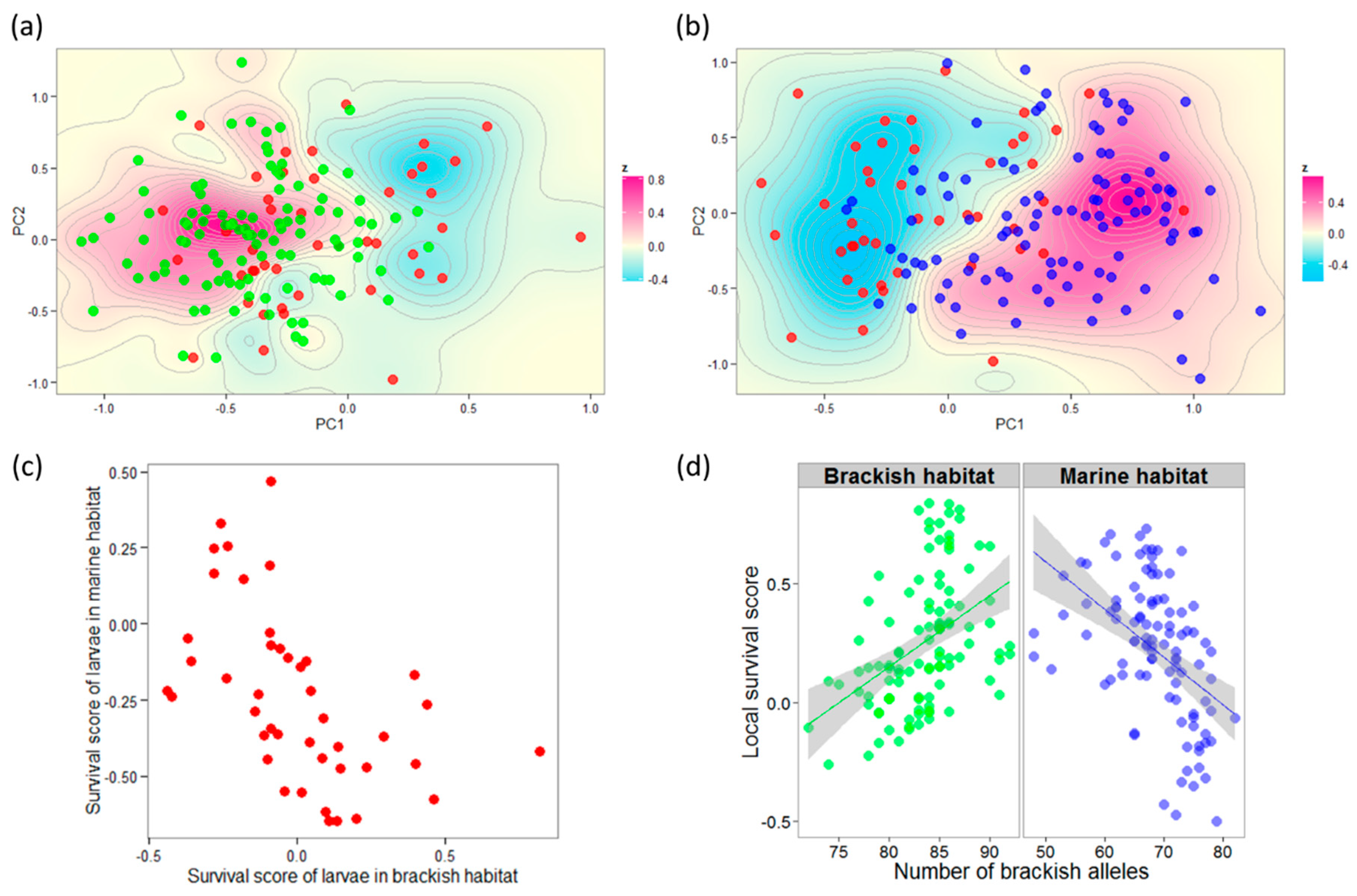
3.3. Genotype–Fitness Links
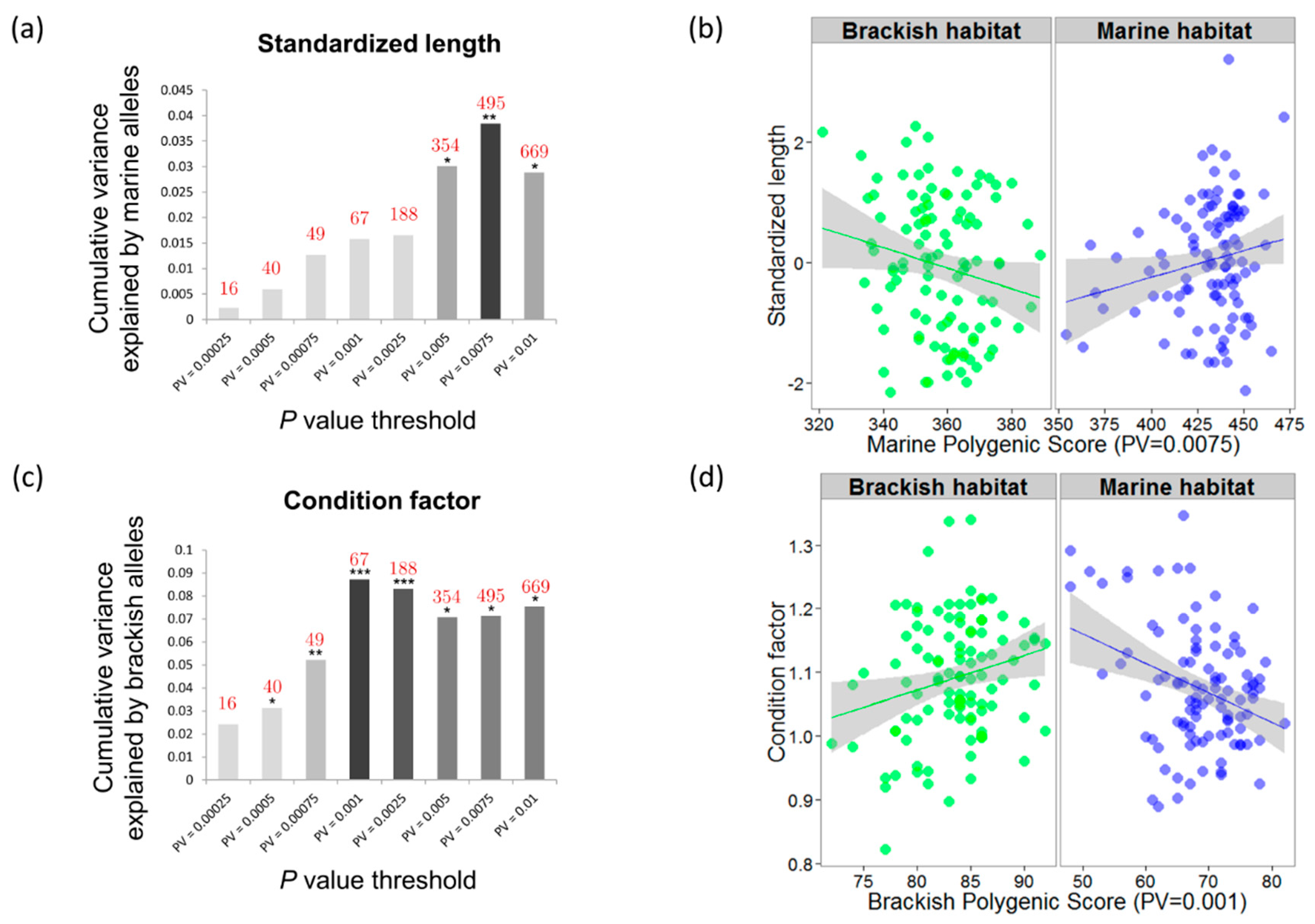
3.4. Genotype–Phenotype Links
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Levene, H. Genetic Equilibrium When More Than One Ecological Niche is Available. Am. Nat. 1953, 87, 331–333. [Google Scholar] [CrossRef]
- Levins, R. Evolution in Changing Environments: Some Theoretical Explorations; Princeton University Press: Princeton, NJ, USA, 1968. [Google Scholar]
- Lenormand, T. Gene flow and the limits to natural selection. Trends Ecol. Evol. 2002, 17, 183–189. [Google Scholar] [CrossRef]
- Hedrick, P.W. Genetic Polymorphism in Heterogeneous Environments: The Age of Genomics. Annu. Rev. Ecol. Evol. Syst. 2006, 37, 67–93. [Google Scholar] [CrossRef] [Green Version]
- Chevin, L.-M.; Lande, R.; Mace, G.M. Adaptation, plasticity, and extinction in a changing environment: Towards a predictive theory. PLoS Boil. 2010, 8, e1000357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sgrò, C.M.; Lowe, A.; Hoffmann, A.A. Building evolutionary resilience for conserving biodiversity under climate change. Evol. Appl. 2010, 4, 326–337. [Google Scholar] [CrossRef] [PubMed]
- Funk, W.C.; McKay, J.K.; Hohenlohe, P.A.; Allendorf, F.W. Harnessing genomics for delineating conservation units. Trends Ecol. Evol. 2012, 27, 489–496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harrisson, K.A.; Pavlova, A.; Telonis-Scott, M.; Sunnucks, P. Using genomics to characterize evolutionary potential for conservation of wild populations. Evol. Appl. 2014, 7, 1008–1025. [Google Scholar] [CrossRef]
- Savolainen, O.; Lascoux, M.; Merilä, J. Ecological genomics of local adaptation. Nat. Rev. Genet. 2013, 14, 807–820. [Google Scholar] [CrossRef]
- Grummer, J.A.; Beheregaray, L.B.; Bernatchez, L.; Hand, B.; Luikart, G.; Narum, S.R.; Taylor, E.B. Aquatic Landscape Genomics and Environmental Effects on Genetic Variation. Trends Ecol. Evol. 2019, 34, 641–654. [Google Scholar] [CrossRef]
- Barrett, R.D.H.; Hoekstra, H.E. Molecular spandrels: Tests of adaptation at the genetic level. Nat. Rev. Genet. 2011, 12, 767–780. [Google Scholar] [CrossRef]
- Wellenreuther, M.; Hansson, B. Detecting Polygenic Evolution: Problems, Pitfalls, and Promises. Trends Genet. 2016, 32, 155–164. [Google Scholar] [CrossRef] [PubMed]
- Pritchard, J.K.; Di Rienzo, A. Adaptation – not by sweeps alone. Nat. Rev. Genet. 2010, 11, 665–667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rockman, M.V. THE QTN PROGRAM AND THE ALLELES THAT MATTER FOR EVOLUTION: ALL THAT’S GOLD DOES NOT GLITTER. Evolution 2011, 66, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Goddard, M.; Hayes, B. Mapping genes for complex traits in domestic animals and their use in breeding programmes. Nat. Rev. Genet. 2009, 10, 381–391. [Google Scholar] [CrossRef]
- De Villemereuil, P.; Frichot, É.; Bazin, E.; François, O.; Gaggiotti, O.E. Genome scan methods against more complex models: When and how much should we trust them? Mol. Ecol. 2014, 23, 2006–2019. [Google Scholar] [CrossRef] [Green Version]
- Hoellinger, I.; Pennings, P.S.; Hermisson, J. Polygenic adaptation: From sweeps to subtle frequency shifts. PLoS Genet. 2019, 15, e1008035. [Google Scholar]
- Yeaman, S. Local Adaptation by Alleles of Small Effect. Am. Nat. 2015, 186, S74–S89. [Google Scholar] [CrossRef]
- Gagnaire, P.-A.; Gaggiotti, O.E. Detecting polygenic selection in marine populations by combining population genomics and quantitative genetics approaches. Curr. Zool. 2016, 62, 603–616. [Google Scholar] [CrossRef] [Green Version]
- Turelli, M.; Barton, N.H. Polygenic Variation Maintained by Balancing Selection: Pleiotropy, Sex-Dependent Allelic Effects and G × E Interactions. Genetics 2004, 166, 1053–1079. [Google Scholar] [CrossRef] [Green Version]
- Gillespie, J.H.; Turelli, M. Genotype-Environment Interactions and the Maintenance of Polygenic Variation. Genetics 1989, 121, 129–138. [Google Scholar]
- Yeaman, S.; Whitlock, M. THE GENETIC ARCHITECTURE OF ADAPTATION UNDER MIGRATION-SELECTION BALANCE. Evolution 2011, 65, 1897–1911. [Google Scholar] [CrossRef] [PubMed]
- Hancock, A.M.; Brachi, B.; Faure, N.; Horton, M.W.; Jarymowycz, L.B.; Sperone, F.G.; Toomajian, C.; Roux, F.; Bergelson, J. Adaptation to Climate Across the Arabidopsis thaliana Genome. Science 2011, 334, 83–86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arnegard, M.E.; McGee, M.D.; Matthews, B.; Marchinko, K.B.; Conte, G.L.; Kabir, S.; Bedford, N.; Bergek, S.; Chan, Y.F.; Jones, F.; et al. Genetics of ecological divergence during speciation. Nature 2014, 511, 307–311. [Google Scholar] [CrossRef] [PubMed]
- Therkildsen, N.O.; Wilder, A.P.; Conover, D.O.; Munch, S.B.; Baumann, H.; Palumbi, S.R. Contrasting genomic shifts underlie parallel phenotypic evolution in response to fishing. Science 2019, 365, 487–490. [Google Scholar] [CrossRef]
- Isnard, E.; Tournois, J.; McKenzie, D.R.; Ferraton, F.; Bodin, N.; Aliaume, C.; Darnaude, A.M. Getting a Good Start in Life? A Comparative Analysis of the Quality of Lagoons as Juvenile Habitats for the Gilthead Seabream Sparus aurata in the Gulf of Lions. Chesap. Sci. 2015, 38, 1937–1950. [Google Scholar] [CrossRef]
- Mercier, L.; Mouillot, D.; Bruguier, O.; Vigliola, L.; Darnaude, A.M. Multi-element otolith fingerprints unravel sea−lagoon lifetime migrations of gilthead sea bream Sparus aurata. Mar. Ecol. Prog. Ser. 2012, 444, 175–194. [Google Scholar] [CrossRef] [Green Version]
- Tournois, J.; Darnaude, A.M.; Ferraton, F.; Aliaume, C.; Mercier, L.; McKenzie, D.R. Lagoon nurseries make a major contribution to adult populations of a highly prized coastal fish. Limnol. Oceanogr. 2017, 62, 1219–1233. [Google Scholar] [CrossRef]
- Pérez-Ruzafa, Á.; Marcos, C.; Perez-Ruzafa, I.M.; Pérez-Marcos, M. Coastal lagoons: “transitional ecosystems” between transitional and coastal waters. J. Coast. Conserv. 2010, 15, 369–392. [Google Scholar]
- Chaoui, L.; Gagnaire, P.-A.; Guinand, B.; Quignard, J.-P.; Tsigenopoulos, C.S.; Kara, M.H.; Bonhomme, F. Microsatellite length variation in candidate genes correlates with habitat in the gilthead sea bream Sparus aurata. Mol. Ecol. 2012, 21, 5497–5511. [Google Scholar] [CrossRef]
- Guinand, B.; Chauvel, C.; Lechene, M.; Tournois, J.; Tsigenopoulos, C.S.; Darnaude, A.; McKenzie, D.; Gagnaire, P. Candidate gene variation in gilthead sea bream reveals complex spatiotemporal selection patterns between marine and lagoon habitats. Mar. Ecol. Prog. Ser. 2016, 558, 115–127. [Google Scholar] [CrossRef] [Green Version]
- Bolger, T.; Connolly, P.L. The selection of suitable indices for the measurement and analysis of fish condition. J. Fish Boil. 1989, 34, 171–182. [Google Scholar] [CrossRef]
- Le Cren, E.D. The Length-Weight Relationship and Seasonal Cycle in Gonad Weight and Condition in the Perch (Perca fluviatilis). J. Anim. Ecol. 1951, 20, 201. [Google Scholar] [CrossRef] [Green Version]
- Rohlf, F.J. tpsDig version 2.17, Department of Ecology and Evolution; State University of New York at Stony Brook: New York, NY, USA, 2013. [Google Scholar]
- Adams, D.C.; Otarola-Castillo, E. geomorph: An r package for the collection and analysis of geometric morphometric shape data. Methods Ecol. Evol. 2013, 4, 393–399. [Google Scholar] [CrossRef]
- Oksanen, J.; Blanchet, F.G.; Kindt, R.; Legendre, P.; Minchin, P.R.; O’Hara, R.; Simpson, G.L.; Solymos, P.; Stevens, M.H.H.; Wagner, H. vegan: Community Ecology Package, version 2.3. 2015. [Google Scholar]
- Baird, N.A.; Etter, P.D.; Atwood, T.S.; Currey, M.C.; Shiver, A.L.; Lewis, Z.; Selker, E.U.; Cresko, W.A.; Johnson, E. Rapid SNP Discovery and Genetic Mapping Using Sequenced RAD Markers. PLOS ONE 2008, 3, e3376. [Google Scholar] [CrossRef] [PubMed]
- Etter, P.D.; Bassham, S.; Hohenlohe, P.A.; Johnson, E.; Cresko, W.A. SNP Discovery and Genotyping for Evolutionary Genetics Using RAD Sequencing. In Methods in Molecular Biology; Springer Science and Business Media LLC, 2011; Volume 772, pp. 157–178. [Google Scholar]
- Catchen, J.; Hohenlohe, P.A.; Bassham, S.; Amores, A.; Cresko, W.A. Stacks: An analysis tool set for population genomics. Mol. Ecol. 2013, 22, 3124–3140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Catchen, J.M.; Amores, A.; Hohenlohe, P.; Cresko, W.A.; Postlethwait, J.H. Stacks: Building and Genotyping Loci De Novo From Short-Read Sequences. G3 Genes|Genomes|Genetics 2011, 1, 171–182. [Google Scholar] [CrossRef] [Green Version]
- Purcell, S.M.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.; Bender, D.; Maller, J.; Sklar, P.; De Bakker, P.I.W.; Daly, M.J.; et al. PLINK: A Tool Set for Whole-Genome Association and Population-Based Linkage Analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef] [Green Version]
- Waples, R.S. Testing for Hardy–Weinberg Proportions: Have We Lost the Plot? J. Hered. 2014, 106, 1–19. [Google Scholar] [CrossRef] [Green Version]
- Jombart, T. adegenet: A R package for the multivariate analysis of genetic markers. Bioinformatics 2008, 24, 1403–1405. [Google Scholar] [CrossRef] [Green Version]
- Gompert, Z.; Comeault, A.A.; Farkas, T.E.; Feder, J.L.; Parchman, T.L.; Buerkle, C.A.; Nosil, P. Experimental evidence for ecological selection on genome variation in the wild. Ecol. Lett. 2013, 17, 369–379. [Google Scholar] [CrossRef] [PubMed]
- Gotthard, K. Growth strategies of ectothermic animals in temperate environments. Environ. Anim. Dev. 2001, 287–304. [Google Scholar]
- Dupont-Nivet, M.; Vandeputte, M.; Vergnet, A.; Merdy, O.; Haffray, P.; Chavanne, H.; Chatain, B. Heritabilities and GxE interactions for growth in the European sea bass (Dicentrarchus labrax L.) using a marker-based pedigree. Aquaculture 2008, 275, 81–87. [Google Scholar] [CrossRef] [Green Version]
- Fonseca, V.; Cabral, H.N. Are fish early growth and condition patterns related to life-history strategies? Rev. Fish Boil. Fish. 2007, 17, 545–564. [Google Scholar] [CrossRef]
- Antonello, J.; Massault, C.; Franch, R.; Haley, C.S.; Pellizzari, C.; Bovo, G.; Patarnello, T.; De Koning, D.-J.; Bargelloni, L. Estimates of heritability and genetic correlation for body length and resistance to fish pasteurellosis in the gilthead sea bream (Sparus aurata L.). Aquaculture 2009, 298, 29–35. [Google Scholar] [CrossRef]
- Navarro, A.; Zamorano, M.J.; Hildebrandt, S.; Ginés, R.; Aguilera, C.; Afonso, J.M. Estimates of heritabilities and genetic correlations for growth and carcass traits in gilthead seabream (Sparus auratus L.), under industrial conditions. Aquaculture 2009, 289, 225–230. [Google Scholar] [CrossRef]
- Saltz, J.B.; Hessel, F.C.; Kelly, M.W.; Information, P.E.K.F.C. Trait Correlations in the Genomics Era. Trends Ecol. Evol. 2017, 32, 279–290. [Google Scholar] [CrossRef]
- Sgrò, C.M.; Hoffmann, A.A.; Sgr, C.M. Genetic correlations, tradeoffs and environmental variation. Heredity 2004, 93, 241–248. [Google Scholar] [CrossRef] [Green Version]
- Le Corre, V.; Kremer, A. The genetic differentiation at quantitative trait loci under local adaptation. Mol. Ecol. 2012, 21, 1548–1566. [Google Scholar] [CrossRef]
- Tournois, J.; Ferraton, F.; Velez, L.; McKenzie, D.R.; Aliaume, C.; Mercier, L.; Darnaude, A.M. Temporal stability of otolith elemental fingerprints discriminates among lagoon nursery habitats. Estuarine Coast. Shelf Sci. 2013, 131, 182–193. [Google Scholar] [CrossRef]
- Ravigné, V.; Dieckmann, U.; Olivieri, I.; Ravignã©V. Live Where You Thrive: Joint Evolution of Habitat Choice and Local Adaptation Facilitates Specialization and Promotes Diversity. Am. Nat. 2009, 174, E141–E169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Purcell, S.M.; Wray, N.R.; Stone, J.L.; Visscher, P.M.; O’Donovan, M.C.; Sullivan, P.F.; Sklar, P.; Ruderfer, D.M.; McQuillin, A.; Morris, D.W. Common polygenic variation contributes to risk of schizophrenia and bipolar disorder. Nature 2009, 460, 748–752. [Google Scholar] [PubMed]
- Speliotes, E.K.; Willer, C.J.; Berndt, S.I.; Monda, K.L.; Thorleifsson, G.; Jackson, A.U.; Allen, H.L.; Lin, D.; Luan, J.; Magi, R.; et al. Association analyses of 249,796 individuals reveal 18 new loci associated with body mass index. Nat. Genet. 2010, 42, 937–948. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sunday, J.M.; Calosi, P.; Dupont, S.; Munday, P.L.; Stillman, J.H.; Reusch, T.B.H. Evolution in an acidifying ocean. Trends Ecol. Evol. 2014, 29, 117–125. [Google Scholar] [CrossRef] [PubMed]
- Hughes, K.A.; Leips, J. Pleiotropy, constraint, and modularity in the evolution of life histories: Insights from genomic analyses. Ann. N. Y. Acad. Sci. 2016, 1389, 76–91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ali, M.; Nicieza, A.G.; Wooton, R. Compensatory growth in fishes: A response to growth depression. Fish Fish. 2003, 4, 147–190. [Google Scholar] [CrossRef]
- Tiffin, P.; Ross-Ibarra, J. Advances and limits of using population genetics to understand local adaptation. Trends Ecol. Evol. 2014, 29, 673–680. [Google Scholar] [CrossRef]
- Hoban, S.; Kelley, J.L.; Lotterhos, K.E.; Antolin, M.F.; Bradburd, G.; Lowry, D.B.; Poss, M.L.; Reed, L.K.; Storfer, A.; Whitlock, M. Finding the Genomic Basis of Local Adaptation: Pitfalls, Practical Solutions, and Future Directions. Am. Nat. 2016, 188, 379–397. [Google Scholar] [CrossRef] [Green Version]
- Lowry, D.B.; Hoban, S.; Kelley, J.L.; Lotterhos, K.E.; Reed, L.K.; Antolin, M.F.; Storfer, A. Breaking RAD: An evaluation of the utility of restriction site-associated DNA sequencing for genome scans of adaptation. Mol. Ecol. Resour. 2017, 17, 142–152. [Google Scholar] [CrossRef]
- Babin, C.; Gagnaire, P.-A.; A Pavey, S.; Bernatchez, L. RAD-Seq Reveals Patterns of Additive Polygenic Variation Caused by Spatially-Varying Selection in the American Eel (Anguilla rostrata). Genome Boil. Evol. 2017, 9, 2974–2986. [Google Scholar] [CrossRef] [Green Version]
- Perrier, C.; Del Campo, A.L.; Szulkin, M.; Demeyrier, V.; Gregoire, A.; Charmantier, A. Great tits and the city: Distribution of genomic diversity and gene-environment associations along an urbanization gradient. Evol. Appl. 2017, 11, 593–613. [Google Scholar] [CrossRef] [PubMed] [Green Version]





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rey, C.; Darnaude, A.; Ferraton, F.; Guinand, B.; Bonhomme, F.; Bierne, N.; Gagnaire, P.-A. Within-Generation Polygenic Selection Shapes Fitness-Related Traits across Environments in Juvenile Sea Bream. Genes 2020, 11, 398. https://doi.org/10.3390/genes11040398
Rey C, Darnaude A, Ferraton F, Guinand B, Bonhomme F, Bierne N, Gagnaire P-A. Within-Generation Polygenic Selection Shapes Fitness-Related Traits across Environments in Juvenile Sea Bream. Genes. 2020; 11(4):398. https://doi.org/10.3390/genes11040398
Chicago/Turabian StyleRey, Carine, Audrey Darnaude, Franck Ferraton, Bruno Guinand, François Bonhomme, Nicolas Bierne, and Pierre-Alexandre Gagnaire. 2020. "Within-Generation Polygenic Selection Shapes Fitness-Related Traits across Environments in Juvenile Sea Bream" Genes 11, no. 4: 398. https://doi.org/10.3390/genes11040398