


 , Na
, Na and
and 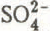 ) were determined by ion chromatography (except H
) were determined by ion chromatography (except HIntroduction
Recent glaciochemical studies provide a better understanding, of the chemical composition of Antarctic precipitation. Most have been devoted to central Antarctic sites such as South Pole (Reference HerronHerron 1982, Reference Delmas, Briat and LegrandDelmas and others 1982[a], Reference Legrand and DelmasLegrand and Delmas 1984) Dome C (Reference Delmas, Barnola and LegrandDelmas and others 1982[b]), Vostok (Reference De Angelis, Legrand, Petit, Barkov, Korotkevich Ye and KotlyakovDe Angelis and others 1984) and Byrd stations (Reference HerronHerron 1982, Reference Palais and LegrandPalais and Legrand 1985).
It has been established that the ionic budget of recently deposited Antarctic snow is mainly contributed by sea-salt aerosol and gas-derived secondary aerosol composed of the three mineral acids H2SO4, HNO3 and HCl. For central areas, it has been clearly demonstrated that the acid contribution dominates. In order to obtain more information about the sources of these acids, it seemed of interest to study the chemistry of snow around the central Antarctic plateau, at sites located between sea-level and ~3000 m elevation. The traverse made in Terre Adélie during the austral summer 1982–83 from Dumont d’Urville to Dome C presented an excellent opportunity to sample recent snow layers at various locations in a relatively homogeneous geographical area. Moreover deeper firn samples were available at two sites (D 55 and D 80) in particular for investigating the influence of the major volcanic eruption of Mt Agung (in 1963) which markedly disturbed atmospheric chemistry on a global scale.
At D 57 (Reference Zanolini, Delmas and LegrandZanolini and others 1985), Vostok (Reference De Angelis, Legrand, Petit, Barkov, Korotkevich Ye and KotlyakovDe Angelis and others 1984), Dome C (Reference Delmas, Barnola and LegrandDelmas and others 1982[b]) and South Pole (Reference Legrand and DelmasLegrand and Delmas 1984), the same analytical procedure had been used to study the chemistry of a wide variety of snow and ice samples, so that it was possible to compare the results found in Terre Adélie to the general features of Antarctic snow chemistry.
Sampling
Eighteen samples of snow were collected during the 1982–83 traverse at 16 different virgin sites between Cap Prud’homme (66°41'S, 139°55'E, near the sea) and D 80 (70°S, 135°E, elevation 2430 m at 433 km from the coast) along the Dumont d’Urville-Vostok axis (Fig.1). The shallow layer (the top 40 cm) was collected directly by pushing PTFE tubes (40 cm long) into snow upwind of the traverse track. At D 80, two samples were recovered in order to get an estimate of the natural reworking of the annual layers by wind. The time intervals integrated by this sampling (from one year near the coast to nearly three at D 55) depend on the local snow accumulation rates. Stringent precautions were taken to avoid contamination of the samples by the operators. All sampling equipment, plastic gloves and tubes had previously been cleaned several times in the laboratory with ultrapure water in a clean room and packed in sealed plastic bags. A blank value related to the sampling procedure was obtained by analyzing ultrapure water poured into PTFE tubes brought back empty to the laboratory.

Fig. 1. Cross-section of the Antarctic ice sheet in the region of Terre Adélie. The positions of the study sites are indicated.
At Dome C (75°S, 124°E, elevation 3250 m, 1070 km from the coast) and D 55 (elevation 2028 m, 200 km from the coast) several hundred samples were collected in hand-dug snow-pits and placed in pre-cleaned disposable 30 ml polystyrene vials (Coulter “Accuvettes”) (at Dome C) or in sealed plastic bags (at D 55). The operators wore clean-room clothing, plastic gloves and masks. Only one part of these sample sets corresponding to the time period from 1959 to 1969 was used for this study. Moreover, an additional set of samples also corresponding to the same time period was obtained by re-coring a firn core recovered at D 80 in a cold room under clean-air conditions. The experimental procedure is described elsewhere (Reference De Angelis, Legrand, Petit, Barkov, Korotkevich Ye and KotlyakovLegrand and others 1984). At Dome C, D 55 and D 80, each individual sample corresponds to about 0.1, 0.1 and 0.3 a, respectively.
Analytical Procedure
All ions with the exception of H+, which was determined by acid titration (Reference Legrand, Aristarain and DelmasLegrand and others 1982), were measured by ion chromatography, using a Dionex apparatus, Model 10 (Reference De Angelis, Legrand, Petit, Barkov, Korotkevich Ye and KotlyakovLegrand and others 1984). Analytical accuracy was typically from 5 to 10% (at 95% confidence limits). Blank values for all ions were found to be negligible with the sampling procedure used for surface samples (PTFE tubes), for the snow-pit at Dome C (Coulter Accuvettes) as well as for the re-cored firn from D 80).
On the other hand, we observed major contamination of the snow collected in the D 55 pit. This contamination, which is only significant for the acidity values, seems to be due to an organic acid contaminant. So far, the actual cause of this effect is not clearly understood (possibly the solvent of the ink in the felt pen used to mark the sample bags). Our analytical procedure also allows us to conclude that F− and HCOO− concentrations are always lower than the detection limits of 0.1 and 0.15 μeq 1−1, respectively. The calculation made on the composition of the acidity (see further in text) confirms that weak organic acids, if present in Antarctic snows, are very minor impurities.
Dating of the Snow and Firn Layers
Dating of the shallow snow samples is not accurate since it was only estimated from the mean value of the accumulation rates at the different sites studied. On the other hand the datings of the D 80 firn core and of the snow-pit samples were made by the method of artificial radioactivity horizons (total beta-radioactivity measurements (Pourchet and Pinglot personal communication)) and is therefore defined with an accuracy of better than 1 a, particularly around the reference horizon of January 1965.
Results and Discussion
The results of the chemical analysis of shallow snow samples are given in Table I and Figure 2. They show that the concentrations of the elements derived from sea salt (Na+, K+ and Cl−) decrease rapidly in the first 100 km with the deposition of the coarsest sea-salt particles. This effect has already been described by Reference Briat, Boutron and LoriusBriat and others (1974) and by Reference Delmas, Briat and LegrandDelmas and others (1982[a]). It seems that the critical parameter for the transport of the bulk of sea-salt aerosol is elevation rather than distance from the coast, as suggested by measurements on the Ross Ice Shelf (Reference Herron and LangwayHerron and Langway 1979). Between D 40 and D 50 elevation increases from 850 to 1730 m. The barrier to the free penetration of sea-salt particles towards the interior probably lies between these two stations.

Fig. 2. Ionic concentrations in surface snow along the study axis in Terre Adélie. Distances (in km) are from Dumont d’Urville. Excess sulphate (
Table I. Ionic Concentrations (in μeq 1−1) in Surface Snow at all Study Stations in Terre adélie (at D80 (1) and (2) correspond to two different samplings. Σ is the sum of ionic concentrations, C the imbalance of the ion budget ((+) indicates an excess of cations vs anions), ![]() and Cl–
ex are excess sulphate and chloride respectively. “H+ calculated” (last column) is the sum
and Cl–
ex are excess sulphate and chloride respectively. “H+ calculated” (last column) is the sum ![]() (see text).)
(see text).)

Such a barrier does not exist for gas-derived secondary aerosol as shown by the results of excess ![]() ,
, ![]() ,
, ![]() and acidity. Excess
and acidity. Excess ![]() and
and ![]() remain in the same concentration range all along the 400 km of the traverse whereas the nitrate content doubles from ~0.5 to −1.0 µeq 1−1 from the coast to the most interior station. Acidity does not exhibit any clear trend as a function of the distance from the coast.
remain in the same concentration range all along the 400 km of the traverse whereas the nitrate content doubles from ~0.5 to −1.0 µeq 1−1 from the coast to the most interior station. Acidity does not exhibit any clear trend as a function of the distance from the coast.
The sulphate values obtained in this work are significantly lower than those reported by Reference Delmas and BoutronDelmas and Boutron (1978). A possible explanation for this discrepancy, apart from the different analytical methods used in these two studies, could lie in the different time periods sampled (1981–82 and 1972–73, respectively) as well as in the representativity of the sampling itself. We have also examined the possible fractionation of sea salt (NaCl) as a function of the distance from the coast. The Cl/Na weight ratios are very close (1.83±0.14) to the bulk sea-water ratio (1.8) along the first 200 km and then increase slightly (Fig. 2). This finding disagrees markedly with the previous values published by Reference Briat, Boutron and LoriusBriat and others (1974) who observed very high values of this ratio further inland (more than 13 at D 80). We think that these authors obtained chlorine concentrations that were much too high (107±44 ng g−1), probably because of contamination of their samples during transportation or storage. Such cases of sample contamination by gaseous chlorine compounds (CH3Cl ?) have been reported previously by us (Reference De Angelis, Legrand, Petit, Barkov, Korotkevich Ye and KotlyakovLegrand and others 1984).
Finally the fractionation factor of potassium with respect to its marine source is found to be close to 1 (0.8) and the correlation between Na and K concentrations (calculated over 17 cases) is excellent (r = 0.994). These findings confirm that potassium has essentially a marine origin in Terre Adélie as already pointed out by Reference BoutronBoutron (1979).
The ionic budget of snow in this geographical area was established by calculating the sum Σ of the ionic concentrations (in µeq 1−1):

Mg2+ and Ca2+ concentrations, which were measured on three of the samples only, were generally estimated from [Na+] values using the ratios of these elements in sea-water. The values of Σ are higher than 10 at the two first stations (D 10 and D 24) only (Table I). The imbalance between cations and anions is always positive (more cations than anions) but remains slight. The highest imbalance is 12% of Σ, a result which is very satisfactory when considering the number of analyses involved in the determination of Σ.
Acidity Composition
As proposed by Reference Legrand and DelmasLegrand and Delmas (1984), the composition of the acidity may be investigated by calculating the sum ![]() and comparing the values obtained to the measured acidities [H+], Excess sulphate and excess chloride are calculated from [Na+]:
and comparing the values obtained to the measured acidities [H+], Excess sulphate and excess chloride are calculated from [Na+]:
Let us compare the measured (8th column of Table I) to the calculated (last column) acidity values. There is a general agreement between the average values and the variations of both parameters. This demonstrates that acidity is satisfactorily explained by the sum of the three mineral acids H2SO4, HNO3 and HCl. The proportion of the latter Cl− exc is, however, nearly negligible, especially in the coastal area (Table II). The percentages given in this table also show that in the last part of the traverse, acid contributions amount to about 2/3 of the total ionic budget, HNO3 being the major ionic trace element present in snow.
Table II Chemical Composition of Snow in Coastal Areas (A) and Between 30 and 430 km From the Sea (B). (Average concentrations Σ and C (see Table I) are in µeql−1. Ionic distribution is in % of ionic budget.)

Finally, it must be noted that ![]() concentrations are found to be very low and stable all along the traverse. This observation shows that the degree of neutralization of the acidity is low (as already found at other Antarctic locations), and also that the Southern Ocean is not a source of ammonia, a conclusion which is in agreement with most works on the origin of this gas in the troposphere.
concentrations are found to be very low and stable all along the traverse. This observation shows that the degree of neutralization of the acidity is low (as already found at other Antarctic locations), and also that the Southern Ocean is not a source of ammonia, a conclusion which is in agreement with most works on the origin of this gas in the troposphere.
Table III summarizes the results obtained for the time period from 1959 to 1969 at D 55, D 80, Dome C and South Pole. In the same table we have also reported a few physical parameters of interest in order to obtain a better understanding of the composition of the origin of the acidity (H2SO4, HNO3 and HCl) in Antarctica. First of all, it must be noted that no clear relationship can be found between the contents of these acids and elevation of snow accumulation rate such as those proposed by Reference HerronHerron (1982) for the nitrate and excess sulphate content of polar snows. Marked differences are observed in the composition of the snow acidity at these four sites. The importance of hydrochloric acid is negligible at D 55, as was observed in coastal areas, whereas it represents nearly one third of the acidity at Dome C. This acid is also particularly important at D 80 and at South Pole. Its formation is assumed to follow the reaction:
at the border of the continent or in the first 100 km where sea-salt aerosol is abundant and where we observed Cl/Na ratios lower than the marine ratio (Table II). In this case HCl would be transported inland and deposited preferentially on the Antarctic plateau.
Table III. Mean Composition of the Acidity at Four Antarctic Locations During the Same Time Period From 1959 to 1969. (Accumulation rates are (1) from M Pourchet (private communication) and (2) from Jouzel and others (l983).)

Nitrate concentrations vary in the range from 0.3 to 1.14 μeq 1−1 at D 55 and from 0.5 to 1.7 μeq 1−1 at D 80. This is also the range observed in the D 57 ice core except for some exceptionally high values (Reference Zanolini, Delmas and LegrandZanolini and others 1985). It represents from 35 to 50% of the acidity during the study period, except at Dome C where its level is exceptionally low (0.17 μeq 1−1, which is 6% of acidity). The maximum values of the nitrate profiles at D 55 (Fig.3) and D 80 (Fig.4) are not found in the same years at both stations: the highest levels in 1968–69 at D 55 are not observed at D 80. Conversely the relatively low values from 1955 to 1965 at D 80 do not appear in the D 55 profile. Therefore the deposition patterns of HNO3 in this area appear to be quite different at distances of only 250 km with an elevation difference of 500 m. Finally, even with sufficient sampling (upper part of the D 55 pit) no clear seasonal variations such as those found at South Pole (Reference Legrand and DelmasLegrand and Delmas 1984) are visible.

Fig. 3. Station D 55. Concentration profiles (in ng g−1 and μeq 1−1) of sulphate and nitrate in a snow-pit from 2 to 4 m depth (time period from 1959 to 1969).

Fig. 4. Station D 80. Concentrations (in ng g−1 and μeq 1−1) of H+, ![]() and
and ![]() in firn from 7 to 12 m depth (time period from 1959 to 1969).
in firn from 7 to 12 m depth (time period from 1959 to 1969).
The case of sulphuric acid is different as its concentration in snow depends on the chronology of large volcanic events (Reference Delmas and BoutronDelmas and Boutron 1980). In the absence of eruptions (for instance during the period from 1959–1964), (Table III) the spatial distribution of H2SO4 fallout seems to exhibit less variability than the other two acids as already discussed by Reference DelmasDelmas (1982). The two extreme mean values of H2SO4 (at D 80 and Dome C) have a ratio of only 2.2 whereas those of HNO3 and HCl have a ratio of 8.4 and ~∞ respectively.
The imprint of the Agung eruption is well marked in the years 1965–66 (Figs.3 and 4). The fluctuations of the sulphate background are in the range from 0.5 to 1.0 and from 0.6 to 1.7 μeq at D 80 and D 55 respectively, whereas the highest sulphate values after Agung are 1.8 and 2.2, indicating that the Agung signal is better defined at D 80 than at D 55 where it lasted apparently only one year (1965). In fact the impact of this eruption on the sulphate profiles of Antarctic snow is much more pronounced in central regions such as Dome C or South Pole (Fig.5). At these two stations the sulphate increase starts at the beginning of 1964, i.e. apparently one year before D 55 and D 80. However, it may be argued that this time-lag of one year probably reflects the detectability of Agung’s fallout over the general sulphate background rather than a different time of first arrival of the volcanic products at these sites.

Fig. 5. The Agung signal (in 1965–66) at four Antarctic locations (D 80, D 55, Dome C and South Pole). Note that the sulphate concentration scale (in ng g−1) is identical for all stations.
Identical scales were chosen for the sulphate concentrations in the four profiles of Figure 5 in order to show that the height of the volcanic signal follows the order Dome C > South Pole > D 80 > D 55. It appears that this order is approximately that of the snow accumulation rates at the four stations, with the exception of D 55 and South Pole, which have nearly the same accumulations (8.3 g cm2 a−1) and do not present identical responses to volcanic fallout (Table III). It may be assumed that geographical considerations should probably also be taken into account in evaluating the impact of a volcanic eruption on Antarctic snow chemistry (Reference Delmas, Legrand, Zanolini and AristarainDelmas and others in press).
Conclusion – Sources of Acids
The spatial and temporal variations reported in this paper provide information on the most important parameters capable of a marked influence on the composition of acids in Antarctic snow.
-
(1) The influence of remoteness from marine surfaces is only appreciable for HCI.
-
(2) Equation (4) probably illustrates the formation of particulate Na2SO4, which is deposited rapidly, and of gaseous HCl, which is allowed to travel further inland.
-
(3) The fact that no clear spatial trend appears in the H2SO4 content of snow does not signify that this acid (“excess-sulphate”) is not of marine origin. This in fact signifies that the life-time of H2SO4 in the Antarctic troposphere is sufficiently long to allow a nearly even deposition pattern over the study area.
-
(4) No effect could be detected on HNO3 or HCl profiles after the eruption of Agung, the impact being limited to the concentrations of sulphate. This was also verified for the eruptions of Tambora (1815) and Galunggung (1822) (Reference Zanolini, Delmas and LegrandZanolini and others 1985). The sulphate profiles are disturbed generally for 2 a after the eruption year and more significantly in low accumulation areas of central Antarctica than in coastal regions.
-
(5) Nitrogen compounds (
and ) apparently have no marine source as is also generally accepted at mid-latitudes.
) apparently have no marine source as is also generally accepted at mid-latitudes.
-
(6) Nitric acid profiles exhibit relatively large spatial and temporal variations. These variations are not sufficiently typical to indicate one HNO3 source rather than another. It has been calculated that an important source of atmospheric HNO3 seems to be located in the upper troposphere in the tropics (lightning) (Reference KleyKley 1983). Long-range transport of this acid to the south polar regions with a subsequent fallout at mid-altitude sites in Antarctica (2000–3000 m) is one possibility. Formation of this acid in the Antarctic troposphere (perhaps by aurorae) cannot be ruled out. Nevertheless our results demonstrate clearly that this acid can sometimes be dominant in the acidity composition of Antarctic snow.
Acknowledgements
We are indebted to M Pourchet for sample collection. This research was financed by Terres Australes et Antarctiques Françaises (TAAF) and Centre National de la Recherche Scientifique Programme Interdisciplinaire de Recherche sur l’Environnement (CNRS-PIREN).








